SONAL DESAI, ARCHITA PATEL JA S. Y. GABHE*
C. U. Shah College of Pharmacy, S. N. D. T. Women’s University, Sir Vithaldas Vidya Vihar, Juhu Tara Road, Santacruz (W), Mumbai – 400 049, Mumbai – 400 049, Intia
Corresponding Author: S. Y:
| Date of Accepted | 16-Jan-2011 |
| Date of Revised | 26-Oct-2010 |
| Date of Received | 9-Apr-2009 |
DOI: 10.4103/0250-474X.89762
Abstract
Yksinkertaista isokraattista käänteisfaasista korkean suorituskyvyn nestekromatografiaa käytettiin erottamaan kolme epäpuhtautta, jotka esiintyivät 8-klooriteofylliininäytteessä. Epäpuhtauksien karakterisointiin käytettiin LC-MS:ää. Massaspektritietojen perusteella näiden epäpuhtauksien rakenteet karakterisoitiin 3,7-dihydro-1,3-dimetyyli-1H-puriini-2,6-dioniksi (epäpuhtaus I), 3,7-dihydro-1,3,7-trimetyyli-1H-puriini-2,6-dioniksi (epäpuhtaus II) ja 8-kloori-1,3-dimetyyli-1,3-dimetyyli-2,6(3H,1H)-purinedionin isomeeriksi (epäpuhtaus III).
Avainsanat
8-klooriteofylliini, epäpuhtaus, LC-MS, käänteisfaasi-HPLC, teofylliini
Esittely
Bulk-lääketeollisuus muodostaa kaikkien lääketeollisuuden alojen perustan, koska se on määritellyn laatuisten vaikuttavien farmaseuttisten ainesosien (API) lähde. Suurimpana haasteena irtotavaralääketeollisuudelle on tuottaa halutun laatuista ja puhdasta lopullista lääkettä taloudellisesti. Vaikutusaineiden puhtaus riippuu useista tekijöistä, kuten raaka-aineista, niiden valmistusmenetelmistä ja kiteytys- tai puhdistusprosessin tyypistä. Täysin puhtaiden materiaalien saaminen on kuitenkin lähes mahdotonta, koska niihin pääsee epäpuhtauksia joko valmistuksen, puhdistuksen tai varastoinnin aikana. Epäpuhtaus on mikä tahansa lääkeaineen komponentti (vettä lukuun ottamatta), joka ei ole lääkeaineeksi määritelty kemiallinen kokonaisuus. Kemiallisella synteesillä tuotettujen lääkeaineiden osalta ICH luokittelee epäpuhtaudet kolmeen luokkaan, joita ovat orgaaniset epäpuhtaudet, epäorgaaniset epäpuhtaudet ja liuotinjäämät. Aineessa olevat epäpuhtaudet voivat olla myrkyllisiä, muuttaa aineen fysikaalisia tai kemiallisia ominaisuuksia ja tehdä siitä siten lääkinnällisesti käyttökelvottoman. Epäpuhtaudet voivat lyhentää tuotteen säilyvyysaikaa ja aiheuttaa vaikeuksia formuloinnissa. Siksi epäpuhtauksien valvonnan lisäksi myös epäpuhtauksien kvalifiointi on kriittinen kysymys irtolääketeollisuudelle.
Antihistamiinit, kuten dimenhydrinaatti ja prometatsiiniteoklaatti, ovat laajalti käytettyjä lääkkeitä matkapahoinvoinnin hoidossa. 8-klooriteofylliini, kemiallisesti 8-kloori-1,3-dimetyyli-2,6(1H, 3H)- purinedioni, on välituote, jota käytetään näiden lääkkeiden suolamuodon valmistuksessa. On tärkeää varmistaa dimenhydrinaatin ja prometatsiiniteoklaatin puhtaus ja turvallisuus. Tämän saavuttamiseksi 8-klooriteofylliiniä on saatava mahdollisimman puhtaana ja tunnettu epäpuhtausprofiili.
Kirjallisuuskatsauksesta käy ilmi, että Wadke et al. tutkivat 9-metyyliisoalloksatsiinin ja 3,9-dimetyyliisoalloksatsiinin vuorovaikutuksia 8-klooriteofylliinin kanssa. 8-klooriteofylliiniä käytettiin sisäisenä standardina uraatin määrittämisessä HPLC-menetelmällä. Se määritettiin myös potentiometrisesti. Kloorifenoksamiinihydrokloridin, 8-klooriteofylliinin ja kofeiinin samanaikainen määritys monikomponenttiannosmuodossa tehtiin ohutkerroskromatografia-densitometrisellä menetelmällä. Gil et al. tutkivat 8-klooriteofylliinin elektroanalyyttistä käyttäytymistä syklisellä voltammetrialla ja differentiaalipulssipolarografialla ja määrittivät sen pitoisuuden lääkevalmisteissa differentiaalipulssipolarografialla. Stabiilisuutta osoittava RP-HPLC-menetelmä kehitettiin ja validoitiin 8-klooriteofylliinille yhdessä difenhydramiinin ja kofeiinin kanssa. Suhteellisuusspektrin nollapisteen ensimmäisen derivaatan spektrofotometrinen ja kemometrinen menetelmä kehitettiin myös kofeiinin, 8-klooriteofylliinin ja kloorifenoksamiinihydrokloridin samanaikaiseen määritykseen ternäärisistä seoksista. Toistaiseksi ei ole raportoitu kromatografista ja spektroskooppista menetelmää 8-klooriteofylliinissä olevien epäpuhtauksien erottamiseksi ja karakterisoimiseksi. Tämän vuoksi tässä työssä pyrittiin eristämään ja karakterisoimaan 8-klooriteofylliinissä esiintyvät epäpuhtaudet käyttämällä nykyaikaisia analyysitekniikoita.
8-klooriteofylliini oli lahjanäyte Kores (India) Ltd:ltä, Thane. Kaikki muut kemikaalit ja reagenssit hankittiin S. D. Fine Chemicals Ltd:ltä (Mumbai, Intia). TLC- ja preparatiivisissa TLC-tutkimuksissa käytetyt liuottimet olivat analyyttistä laatua ja HPLC-tutkimuksissa käytetyt liuottimet HPLC-laatua. Puskurin valmistukseen käytettiin AR-luokan natriumasetaattitrihydraattia.
Aluksi tehtiin TLC-tutkimuksia, jotta tiedettäisiin näytteessä olevien epäpuhtauksien määrä. Stationäärifaasina käytettiin esipinnoitettuja TLC-levyjä, jotka olivat silikageeliä 60GF254 (Merck). Näyte liuotettiin pieneen määrään etyyliasetaattia, ja tätä liuosta käytettiin TLC-levyjen täplittämiseen. Eri liikkuvia faaseja kokeiltiin. Etyyliasetaatti:tolueeni:jääetikkahappo (10:0,3:0,5 v/v/v) erotti paremmin kuin muut liikkuvat faasit. TLC:n avulla 8-klooriteofylliininäytteestä erotettiin neljä komponenttia, joiden Rf-arvot olivat 0,029, 0,132, 0,198 ja 0,852. 8-klooriteofylliinin Rf-arvo oli 0,852.
Kun TLC:n liikkuva faasi oli kehitetty, seos yritettiin erottaa preparatiivisella TLC:llä. Näyte liuotettiin vähimmäismäärään etyyliasetaattia ja pilkottiin kaistaleiksi. Kaikki kolme epäpuhtautta erotettiin 8-klooriteofylliinistä preparatiivisella TLC:llä käyttäen samoja kromatografisia olosuhteita, joita käytettiin TLC-tutkimuksissa. Eri kaistat kerättiin erikseen ja uutettiin etyyliasetaatilla. Koska kunkin eristetyn epäpuhtauden I, II ja III määrät olivat hyvin pieniä, nämä epäpuhtaudet päätettiin eristää yhdessä. Epäpuhtaudet I, II ja III nimettiin yhdessä fraktioksi I. Fraktio I eristettiin 8-klooriteofylliinistä preparatiivisella TLC:llä. Koska kutakin epäpuhtautta ei eristetty erikseen, erilaisia tunnistustekniikoita, kuten IR- ja NMR-tekniikkaa, ei voitu käyttää. Päätettiin tehdä lisätutkimuksia LC-MS:llä, joka mahdollistaa samanaikaisen erottelun ja karakterisoinnin. Ennen LC-MS-analyysia kehitettiin HPLC-profiili fraktiolle I. HPLC-tutkimuksissa käytettiin Tosohin korkean suorituskyvyn nestekromatografia, joka oli varustettu CCPM-kiertopumpulla, PX8010-pumpunohjaimella ja UV-detektorilla. Injektioventtiiliyksikköön asennettiin 20 µl:n silmukka. Fraktio I liuotettiin asetonitriiliin ja sille tehtiin käänteisfaasi-HPLC-analyysi liikkuvalla faasilla, joka koostui asetonitriilistä: natriumasetaattitrihydraatti (pH 3,57; 0,01 M) (5:95 v/v). Kolonniksi valittiin Phenomenex ODS (250 × 4,6 mm I.D.; hiukkaskoko 5 µm). Virtausnopeus oli 1,5 ml/min ja detektointia seurattiin aallonpituudella 280 nm. Liikkuva faasi suodatettiin sintratun G5-lasisuodattimen läpi tyhjiössä ennen käyttöä ja sonikoitiin ilmakuplien poistamiseksi. Jakeen I HPLC-analyysissä havaittiin myös kolme piikkiä, joiden keskimääräiset retentioajat olivat 8,324 min, 14,275 min ja 20,933 min (kuva 1). Piikki, jonka keskimääräinen retentioaika oli 2,086 min, oli liuotinpiikki.
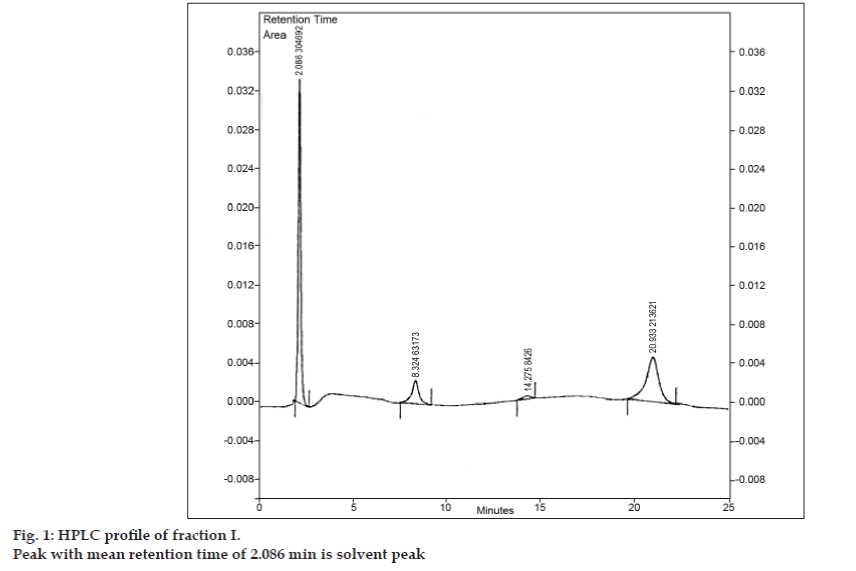
Kuvio 1: Jakeen I HPLC-profiili.
Jakeelle I tehtiin tämän jälkeen LC-MS-analyysi epäpuhtauksien karakterisointia varten. LC-MS-tutkimukset suoritettiin järjestelmällä, jossa LC-osa koostui 1100-sarjan HPLC:stä (Agilent Technologies, USA), joka sisälsi tyhjiökaasunpoistimen (G1322A), kvaternaaripumpun (G1311A), automaattisen näytteenottimen (G1313A) ja UV/näkyvyysilmaisimen (G1314A), ja MS-osa koostui kolmoiskvadrupoli-massaspektrometristä Quattro II:lla (Micromass UK Ltd.),
LC-MS-tutkimuksissa nestekromatografiset erotukset tehtiin Phenomenex C18 -kolonnilla (250 × 4,6 mm, 5 µm) huoneenlämmössä liikkuvassa faasissa, jonka liikkuvana faasina oli asetonitriili: natriumasetaattipuskuri (5:95, v/v) virtausnopeudella 1,5 ml/min. Massaspektrometriä käytettiin positiivisessa elektronisuihkuionisaatiomoodissa (ESI) massa/varaus (m/z) -suhteella välillä 120-500 m/z. Sumutuskaasuna käytettiin typpeä. Tiedot kerättiin ja käsiteltiin Masslynx-ohjelmistolla. Epäpuhtauksista saatiin massaspektritiedot (kuvat 2, 3 ja 4). Kolmen piikin fragmentoitumisreitille on ominaista metyyliryhmän ja/tai karbonyyliryhmän häviäminen. Piikit, joiden m/z-arvot ovat 180,9, 195,7 ja 214,9, vastaavat +, + ja + piikkejä. Saatujen MS-tietojen mukaan 8,324 minuutissa, 14,275 minuutissa ja 20,933 minuutissa eluoituvat epäpuhtaudet olivat vastaavasti teofylliiniä (moolimassa 180), kofeiinia (moolimassa 194) ja 8-klooriteofylliinin isomeeria (moolimassa 214,5) (taulukko 1). Näin erotettiin kolme epäpuhtautta ja niiden rakenteet selvitettiin massaspektritietojen perusteella (kaavio 1).
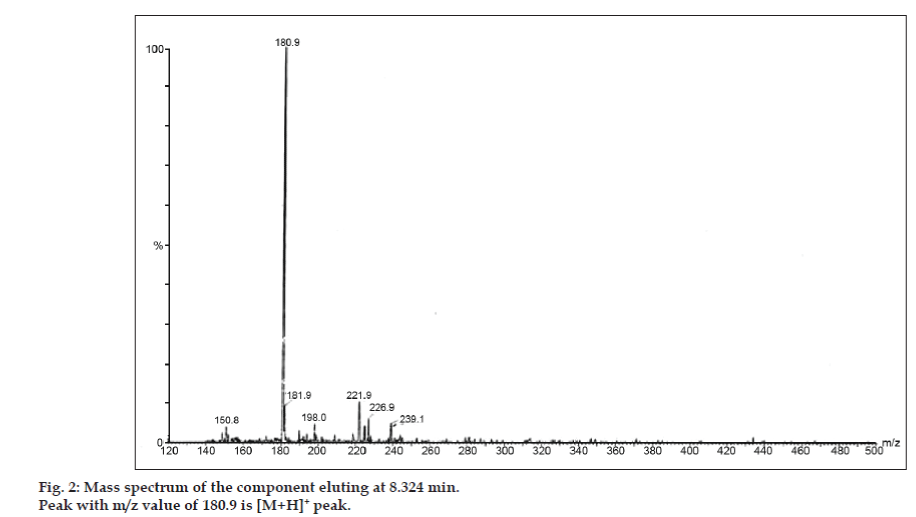
Kuva 2: Komponentin massaspektri, joka eluoitui 8,324 min.
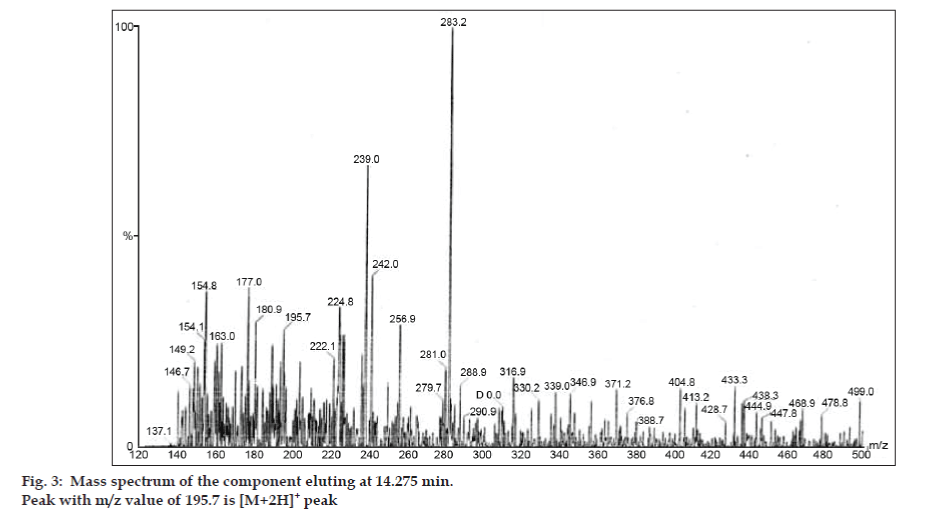
Kuva 2: Komponentin massaspektri. 3: Komponentin massaspektri, joka eluoituu 14.275 min.
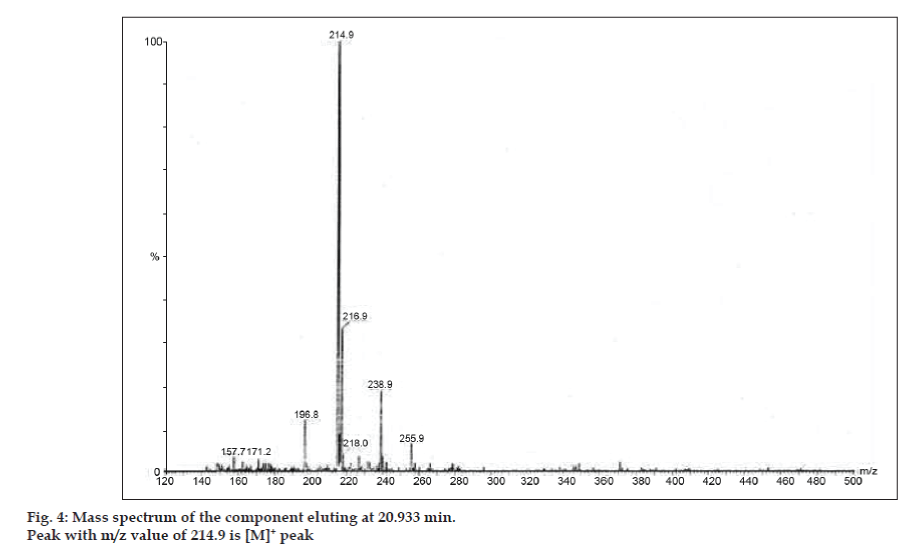
Kuva. 4: Komponentin massaspektri, joka eluoituu 20.933 min.
| Peak no. |
Pidätysaika (min) |
Fragmentti-ionit (m/z) | Tunnistus |
|---|---|---|---|
| 8.324 | 181.9 +, 180.9 +, 150.8 + | Teofylliini | |
| 14.275 | 195.7 +, 180.9 +,149.2 +, 137.1+ | Kofeiini | |
| 20.933 | 216.9 +, 214.9 +, 171.2 +, 157.7 + | 8- klooriteofylliinin isomeeri |
TAULUKKO 1:
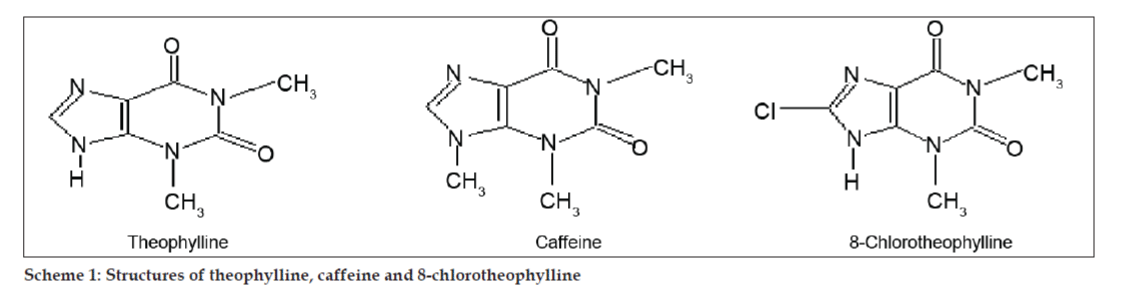
Kaavio 1: Teofylliinin, kofeiinin ja 8-klooriteofylliinin rakenteet
Kiitokset
Tekijät ovat kiitollisia Kores (India) Ltd.:lle, Thane lahjanäytteen antamisesta 8-klooriteofylliinistä.
- Kasture AV, Wadodkar SG, Mahadik KR, More HV. Pharmaceutical Analysis. Vol. 1. Pune: NiraliPrakashan; 1997. s. 12-4.
- United State Pharmacopoeia, Vol. 26. Rockville, MD: United States Pharmacopoeia Convention, Inc.; 1999. s. 2049-59.
- Ahuja S, Alsante KM. Handbook of isolation and characterization of impurities in pharmaceuticals. California: Academic Press; 2003. s. 6.
- Foye WO, Lemke TL, Williams DA. Lääkekemian periaatteet. 4th ed. New Delhi: B. I. Waverly Pvt Ltd; 1995. s. 419.
- Reynolds JE. Martindale-The Extra Pharmacopoeia. 29th ed. London: Pharmaceutical Press; 1989. s. 451,459.
- Wadke DA, Guttman DE. Kompleksin muodostumisen vaikutus reaktionopeuteen III. Joidenkin isoalloksatsiinien vuorovaikutus 8-klooriteofylliinin kanssa spektri-, liukoisuus- ja kineettisin menetelmin määritettynä. J Pharm Sci 1965;54:1293.
- Bennett MJ, Patchett BP, Worthy E. A simple HPLC method for the determination of urate in serum and urine using 8-chlorotheophylline as internal standard. Med Lab Sci 1984;41:108-11.
- Nikolic K, Medenica M. 8-klooriteofylliinin potentiometrinen määritys. Microchimica Acta 1986;88:5.
- Bebawy LI, El-Kousy NM. Joidenkin monikomponenttiannosmuotojen samanaikainen määritys kvantitatiivisella ohutkerroskromatografian densitometrisellä menetelmällä. J Pharm Biomed Anal 1999;20:663-70.
- Gil EP, Blazquez LC, Garcia-MoncoCarra RM, Misiego AS. 8-klooriteofylliinin polarografinen käyttäytyminen ja sen määrittäminen annosmuodoissa. Elektroanalyysi 1993;5:343.
- Barbas C, Garcia A, Saavedra L, Castro M. Kofeiinin, 8-klooriteofylliinin ja difenhydramiinin määritysmenetelmän optimointi ja validointi isokraattisella korkean suorituskyvyn nestekromatografialla Kestävyyden arvioinnin stressitesti. J ChromatogrA 2000;870:97-103.
- Kelani KM. Kofeiinin, 8-kloroteofylliinin ja kloorifenoksamiinihydrokloridin samanaikainen määritys ternäärisistä seoksista suhdelukujen nollapisteen ensimmäisen derivaatan spektrofotometrisin ja kemometrisin menetelmin. J AOAC Int 2005;88:1126-34.
.