Indledning
Rheumatoid arthritis (RA) er en autoimmun sygdom, polygen i naturen, karakteriseret ved polyarthritis med systemiske manifestationer og øget og alvorlig morbiditet.1,2 RA rammer 0,5 %-1 % af befolkningen og medfører en forringelse af livskvaliteten, betydelig fysisk invaliditet og betydelige økonomiske omkostninger.3-6 Det kliniske udtryk for sygdommen er varieret og spænder fra milde selvbegrænsende former til en meget aggressiv, hurtig udvikling, der kulminerer med ødelæggelse af det angrebne led og den deraf følgende invaliditet.7
Genetiske undersøgelser har bekræftet eksistensen af et genetisk substrat, der til dels er relateret til visse gener, der koder for proteiner, der er involveret i T-cellernes respons.1 Disse resultater styrker betydningen af den rolle, der tilskrives T-cellerne i initieringen og opretholdelsen af det unormale immunrespons i denne sygdom.8
Patogenesen af RA er kompleks og involverer forskellige cellepopulationer, der er relateret til det medfødte og adaptive immunrespons. Residente celler i synovium, som f.eks. fibroblastiske synoviocytter B eller makrofager i intimaen, og inflammatoriske celler fra blodet som T-lymfocytter, B-lymfocytter og monocytter9 er involveret i patogenesen. De bidrager alle til den aggressive transformation af synoviocyt B-fænotypen og udviklingen af et intenst inflammatorisk infiltrat med slutresultatet brusk- og subchondral knogledestruktion10,11 (fig. 1).
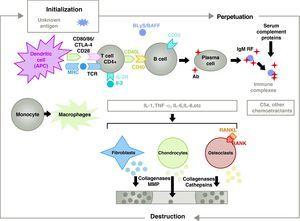
Patofysiologi ved reumatoid arthritis. Generel patofysiologisk organisering af reumatoid arthritis. AC, antistof; BAFF, B-celleaktiverende faktor; BLyS, B-lymfocyt-stimulator; CD, cluster of differentiation; CPA, antigenpræsenterende celle; CPH, MHC; CTLA4, lymfocyt-associeret antigen 4 T cytotoksisk C5a-fraktion komplement 5a, FR, reumatoid faktor; Ig, immunoglobulin; IL, interleukin; MMP, matrixmetalloproteinaser; RANK, receptor activator of nuclear factor B kappa; RANKL, receptor activator ligand for nuclear factor B kappa; RCT, T-celle receptor; TNF, tumor necrosis factor.
Den nuværende behandling af RA er baseret på administration af sygdomsmodificerende antirheumatika (DMARDs), der anvendes alene eller i kombination.12 Disse lægemidler bremser leddestruktionen, dvs. de er i stand til at ændre det naturlige sygdomsforløb.4,13 Procentdelen af patienter med et tilfredsstillende klinisk respons er imidlertid lav og kræver ofte tilføjelse af et biologisk lægemiddel hos en høj procentdel af patienterne.9,13-15
I de seneste år er der blevet identificeret nye molekyler og terapeutiske mål, hvis blokade kan reducere eller fjerne det kroniske inflammatoriske respons. Et af disse nye molekyler er abatacept. Abatacept er en fuldt humaniseret proteinkonstruktion, der består af det ekstracellulære domæne af humant cytotoksisk T-lymfocyt-associeret antigen 4 (CTL4) og et genetisk manipuleret fragment af Fc-regionen af humant immunoglobulin G1 (IgG1), som hæmmer de kostimulerende T-celler, der virker på den egentlige kerne af immunresponset og derfor i sygdommens begyndelse.
T-celleaktivering
Effektiv immunaktivering af T-celler kræver deltagelse af to grupper af membranreceptorer på antigenpræsenterende celler (APC)14 (fig. 1 og 2). Den første er det medium, som APC’erne bruger til at levere det tidligere forarbejdede specifikke antigen til T-cellen. På trods af den enorme indsats, der er lagt i denne forskning, er vi stadig ikke i stand til at identificere de arthritogene antigener, der udløser RA8 . APC’ernes præsentation af et antigen, mod hvilket et specifikt immunrespons er organiseret gennem et trimolekylært kompleks, der omfatter: molekyler af det store histokompatibilitetskompleks (MHC), der er til stede i APC’en, antigenet, mod hvilket immunresponset udvikles, og en membranreceptor på T-cellen (TCR), der er specifik for dette antigen15 (signal eller signalvej for immunresponset 1).
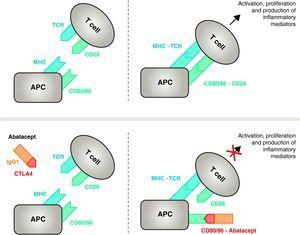
Mekanisme for abatacepts virkning. Abataceptfragmentet, der omfatter det ekstracellulære domæne af CTLA4, binder sig til CD80/CD86-receptorer og forhindrer eller fortrænger dets interaktion med CD28-receptoren. På denne måde blokerer det selektivt den specifikke binding af CD80/CD86 til CD28-receptoren, hvilket patofysiologisk set er en blokering af det andet signal for immunaktivering og dermed aktivering af T-celler CPA, antigenpræsenterende celle; MHC, større histokompatibilitetskompleks; TCR, T-celle-receptor.
For at hæmme fuld aktivering kræver T-cellerne et andet sæt af intercellulære kommunikationsreceptorer mellem APC’er og T-celler, som sker gennem kostimulerende veje og udgør det såkaldte 2-signal-immunrespons14 . Selv om der findes flere kostimulerende veje, er én af dem afgørende, nemlig bindingen af receptorerne CD80 (B7-1)/CD86 (B7-2) på CPA’s membran med CD28-receptoren på T-celler.10,16 Samtidig aktivering af begge udløser intens intracellulær signalering i T-celler, som er afgørende for fuld aktivering, proliferation, overlevelse og cytokinproduktion 8. 24-48 timer efter aktivering af T-lymfocytter igangsætter den samme intracellulære signalering en reguleringsmekanisme, der har til formål at deaktivere selve responsen. Dette inducerer ekspression af CTLA411 på lymfocytcellemembranen med den opgave at konkurrere med CD28 på grund af dens større bindingsaffinitet til CD80/CD86.17,18
Aktiveringen af begge T-celleundergrupper, CD4+ og CD8+, afhænger af den kostimulerende CD28-receptor. CD4+ T-celler er hjælpende T-celler. De genkender de peptider, der præsenteres af MHC-klasse II-molekyler, som findes på APC’en. Disse antigener stammer fra den eksogene vej, der behandler patogener som f.eks. bakterier. Mange autoimmune sygdomme er forbundet med et patologisk respons fra CD4+ T-celler. CD8+ T-celler er for deres vedkommende cytotoksiske lymfocytter (CTL). CD8+ T-celler genkender antigener, hovedsagelig virale og tumorale antigener, der præsenteres af klasse I-molekyler MHC. Ved aktivering formidler CD8+ cellerne ødelæggelse af målceller gennem produktion af perforin, granzymer og interferon (IFN)-g. Begge subtyper af T-celler aktiveres ved kostimulering med CD2815, selv om aktiveringen af CD8+ T-celler er mindre afhængig af denne kostimuleringsvej. Faktisk udtrykker alle CD4+ celler CD28 på deres membran, mens dette kun sker hos ca. 50 % af CD8+ cellerne.19 Desuden har det vist sig, at CD4+ celler udviser et større respons på CD2820-binding. Desuden er CD28-promotoren ikke et absolut krav for aktivering af CTL.21 Alt dette ville give en dobbelt terapeutisk fordel i klinisk praksis. På den ene side virker abatacept fortrinsvis på målcellen i sygdommens patogenese. Desuden ville den reducerede virkning på aktiviteten af CD8+-lymfocytter sikre en bedre sikkerhedsprofil med hensyn til virale og tumorale komplikationer.
Aktivering af CD4+ T-celler er startskuddet til en proinflammatorisk kaskade med produktion af store mængder cytokiner og celleproliferation, der, hvis den fortsætter og opretholdes, som ved RA, fører til en meget aktiv kronisk inflammation, der er i stand til at ødelægge de væv, hvor den udløses, for det meste leddene i tilfælde af RA8 (fig. 1). Synovien begynder at proliferere på grund af infiltrerende celler fra blodet, herunder selve T-lymfocytterne og deres undertyper samt B-lymfocytter Monocytterne differentierer sig til makrofager og osteoklaster og aktiverer også ledkirtelchondrocytter. I dette miljø er der store mængder af proinflammatoriske cytokiner såsom interleukin (IL)-1, IL-6 og tumornekrosefaktor (TNF) og mange andre. B-cellerne producerer også autoantistoffer som f.eks. reumatoid faktor eller anti-citrullinated peptid-antistoffer. Alle disse fører til ødelæggelse ikke kun af synovialmembranen, men også af den underliggende knogle og brusk.22
Bioteknologi i behandlingen af reumatoid arthritis
På grund af ovennævnte forskning er der blevet udviklet og markedsført bioteknologisk fremstilling af forskellige molekyler, der har til formål at blokere specifikke mål. Den første generation var kendetegnet ved fremkomsten af TNF-neutraliserende lægemidler: etanercept, infliximab og adalimumab samt anakinra, som hæmmer virkningen af IL-1. Efterfølgende er der kommet nye molekyler til, f.eks. abatacept, der modulerer kostimuleringen af immunresponset, certolizumab og golimumab, der blokerer TNF, rituximab mod CD20-receptoren for B-lymfocytter og tocilizumab, der blokerer IL-6.7,23-26
Trods det store spring i terapeutisk effektivitet som følge af indførelsen af disse lægemidler reagerer en betydelig procentdel af patienterne, anslået til mellem 25 % og 40 %, ikke på de lægemidler eller biologiske stoffer, der i øjeblikket markedsføres, eller er ramt af bivirkninger27 .32 Behovet for at forbedre denne situation er fortsat en tilskyndelse til at forfølge og udvikle nye molekyler, der har til formål at regulere forskellige terapeutiske mål, som kan forbedre den terapeutiske effektivitet, som f.eks. abatacept, der selektivt modulerer aktiveringen af T-celler33 .
Abatacept er et proteinkonstrukt, der fremstilles ved hjælp af rekombinant DNA-teknologi i hamsterovarieceller.34,35 Dette molekyle blev designet til at gribe ind i reguleringen af kostimulerende veje i T-celler, som spiller en vigtig rolle i patogenesen af forskellige autoimmune sygdomme, infektioner, afstødning af transplanterede organer og tumorimmunitet.36
Abatacept anvendes i kombination med methotrexat til RA-patienter, som har haft et utilstrækkeligt respons eller intolerance over for andre DMARD’er, herunder methotrexat (MTX) eller en TNF-alfa-hæmmer. Ved polyartikulær juvenil idiopatisk arthritis er det indiceret hos patienter på 6 år eller derover, som har haft et utilstrækkeligt respons på andre DMARD’er, herunder mindst ét TNF-neutraliserende lægemiddel35 .
Virkningsmekanisme for Abatacept
Abatacept er en selektiv modulator af det CD80/86-CD28-kostimulerende signal, og som tidligere omtalt er det afgørende for aktivering af T-celler Abatacept hæmmer T-celleaktivering, idet det selektivt blokerer specifik binding af CD80/CD86-receptoren i APC til CD28 på T-cellen (Fig. 2).22,37 Den farmakologiske strategi søger at hæmme det accelererede immun/inflammatoriske respons, der er karakteristisk for sygdommen, og genoprette normal homøostase i immunsystemet. Faktisk er konkurrencen mellem endogene CD28 og CTLA4 om at binde sig til CD80/86 den fysiologiske mekanisme, der anvendes til at regulere og, hvor det er relevant, afslutte et normalt immunforsvar. Abatacept hæmmer ved at blokere bindingen af CD80/86 til CD28 overførslen af et andet signal i immunresponset, som indirekte giver et negativt signal om T-celleaktivering. Desuden har abatacept sandsynligvis en større virkning ved at forhindre dannelsen af et kostimulerende signal i T-celler, idet det inaktiverer de allerede aktive, som ikke er bundet til T-celle CTLA4
Supporting Drug for Use
1. Hvorfor er abatacept medtaget i gruppen af immunmodulerende lægemidler? Dybest set fordi det giver celleudtømning, især af T-celler på grund af den farmakologiske virkning, der udøves ved ikke at blokere selektivt for et bestemt cytokin, hvorved man undgår den radikale undertrykkelse af væsentlige veje for korrekt fungerende immunrespons.8
2. Hvordan forhindrer det bindingen af molekylets Fc-region til dets receptor? Fc-regionen i abatacept er genetisk modificeret, så den ikke binder til CD16- og CD32-receptorerne og kun i meget svag grad til CD64-receptoren. Dette design omgår de cellulære reaktioner, der formidles af Fc-receptoren, såsom antistofafhængig cellulær cytotoksicitet (ADCC) og komplementafhængig cytotoksicitet (CDC).18 Begge er forbundet med cellelysis med potentielle negative virkninger, der kan ses i forbindelse med forlængede38 behandlinger. Derfor synes det modificerede fragment af IgG1 at være aktivt og dermed at forhindre bivirkninger som følge af ADCC.39
3. Antiinflammatorisk virkning af abatacept. Abatacept reducerer betydeligt mange af de inflammatoriske mediatorer hos patienter med RA og genopretter dem til det normale, hvilket er påvist i flere kliniske forsøg, der blev anvendt ved forskningen af lægemidlet.
I en fase II-b, 1-årig, placebokontrolleret undersøgelse hos patienter med RA og utilstrækkelig respons på MTX blev der taget prøver og målt serumniveauer af udvalgte markører i dagene før infusionen for at undersøge abatacepts virkning på mediatorer og proinflammatoriske cytokiner. En gruppe patienter fik MTX og abatacept 10 mg/kg i henhold til et regelmæssigt skema. Kontrolgruppen blev i mellemtiden behandlet med MTX og placebo. Et år efter behandlingen var markørerne i abataceptgruppen på 10mg/kg normaliseret, mens de fortsat var forhøjede i placebogruppen (TNF: 7,4 vs. 10,3pg/ml; FR: 159 vs. 225U/l, sIL-2R: vs. 1228,3. 1697,1pg/ml IL-6: 7,3 vs 19,9pg/ml).40
4. Immunogenicitet. Ifølge data om lægemidlet udviklede kun 187 ud af 3877 (4,8 %) patienter med RA behandlet i op til 8 år med abatacept antistoffer mod lægemidlet under behandlingen.41 Antistoffer mod abatacept blev evalueret hos patienter efter ophør med lægemidlet (>42 dage efter den sidste dosis), og hos 103 ud af 1888 (5,5 %) var seropositive. I en anden undersøgelse af 2000 patienter abatacept blev antistoffer derimod målt i en anden undersøgelse af 2000 patienter abatacept, og det blev konkluderet, at abatacept har lav immunogenicitet.42,43
5. Abatacept og tuberkulose. TNF deltager i det inflammatoriske respons og immunopatologien ved tuberkulose (TB). In vitro-undersøgelser viser, at TNF øger den fagocytiske aktivitet og mykobakteriedræbende makrofager, mens de in vivo var involveret i den indledende dannelse og den efterfølgende vedligeholdelse af granulomer, noget, der kontrollerer væksten af mykobakterier og begrænser dens spredning. I en kronisk model for reaktivering af latent TB hos mus undersøgte vi udviklingen af infektionen hos mus, der blev behandlet med abatacept, sammenlignet med en anden gruppe, der blev behandlet med et murint monoklonalt anti-TNF.42 4 måneder efter at have inficeret C57BL/6-mus med Mycobacterium tuberculosis, og efter at det var blevet bekræftet, at de havde en latent TB-infektion, blev musene behandlet i 16 uger med en af to eksperimentelle interventioner. Efter denne periode døde alle de mus, der blev behandlet med anti-TNF, af dissemineret TB med en gennemsnitlig overlevelse på 44 dage. Derimod døde ingen af de mus, der blev behandlet med abatacept.
Mens koncentrationen af IFN-g i serum ikke ændrede sig i abatacept-gruppen, var den forhøjet hos musene med anti-TNF. Denne stigning blev tilskrevet den øgede infiltration af CD4+ og CD8+ forårsaget af den udbredte spredning af bakteriekolonierne.
Så, mens mus, der blev behandlet med anti-TNF-behandling, viste 100 % dødelighed, ændrede abatacept ikke musenes evne til at organisere et inflammatorisk respons, der var i stand til at kontrollere spredningen af tuberkulose. Der er dog stadig ikke tilstrækkelige kliniske data til at bekræfte disse resultater hos mennesker.
6. Antiresorptiv virkning af abatacept på knogleomdannelse. Osteoklastaktiviteten øges ved RA, både i leddet, hvilket forårsager knogleerosioner, og systemisk, idet den når op på niveauer, der er forbundet med generaliseret osteoporose.44,45
Der er faktisk påvist en stigning af liganden receptoraktivator af kernefaktoren NF-kB (RANKL) i synovialmembranen.45,46 Abatacept hæmmer dosisafhængigt dannelsen af murine osteoklaster og den osteoklastogene aktivitet, der er vurderet in vitro. Dette blev undersøgt i murine osteoklaster dyrket på dentinplader, hvor antallet af resorptionshuller blev målt efter 6 dages tilførsel af forskellige doser abatacept.47
Medikamentet reducerede signifikant arealet af knogleresorption. Disse data tyder på, at abatacept er et molekyle, der binder direkte til osteoklastprækursorcellerne og hæmmer deres differentiering. Denne mekanisme kunne forklare lægemidlets anti-erosive virkning hos patienter med RA. Faktisk viste patienter, der blev behandlet med abatacept, en faldende tendens i RANK- og dets ligand RANKL-niveauer i synovium, der alle var forbundet med øget osteoprotegerin.48 Selv om den nøjagtige mekanisme, der ligger til grund for denne observation, er uklar, korrelerer disse fund godt med den radiologiske forbedring, der blev observeret hos patienter, der blev behandlet med abatacept.
7. Virkninger af abatacept i andre immunceller. Selv om APC er den målcelle, der binder abatacept, og makrofager også udtrykker CD80/86-receptorer på deres overflade, er der kun få undersøgelser, der undersøger lægemidlets virkning på aktiviteten af disse celler. En nyere in vitro-undersøgelse har faktisk vist, at makrofager udviste en markant CD80/86-receptorekspression, og behandling med abatacept reducerede cytokin49 -produktionen betydeligt. Disse resultater tyder på, at lægemidlets virkningsmekanisme kan udvides til at omfatte reguleringen af makrofaglinjen, som er nøgleceller i sygdommens patogenese.
Abatacept undertrykker også den follikulære migration af antigenspecifikke T-celler og dermed samarbejdet mellem T-celler og follikulære B-celler i lymfeknuden. Dette fund er blevet observeret in situ i lymfeknuder hos BALB/c50-mus. Efter transfusion af sådanne mus med antigenspecifikke præstimulerede T-celler viste en efterfølgende immunisering af musene T-celleproliferation og migration til B-lymfocytområdet. Hos mus, der blev behandlet med abatacept, blev T-celleproliferation og -migration blokeret, hvilket begrænsede deres tilstedeværelse i de fleste tilfælde i lymfeknudeparacortexet. Således reducerer langvarig behandling med abatacept proliferation, mobilitet og fordeling af intraganglionar autoantigenhukommelseslymfocytter, hvilket kan føre til et fald i autoantistoffer.
Konklusioner om virkningsmekanismen for Abatacept
Abatacept er en fuldt humaniseret proteinkonstruktion, der består af det ekstracellulære domæne af humant cytotoksisk T-lymfocyt-associeret antigen 4 (CTL4) og et genetisk modificeret fragment af Fc-regionen af IgG1, designet til at gribe ind i reguleringen af T-lymfocytternes kostimulering Lægemidlet hæmmer aktivering af T-celler ved selektivt at blokere den specifikke binding af CD80/CD86 til CD28-receptoren og dermed hæmmer T-celleproliferation og immunreaktioner fra B-lymfocytter Denne farmakologiske virkning resulterer i nedsatte niveauer af inflammatoriske mediatorer hos patienter med RA og i et sikkert og effektivt klinisk respons.
Interessekonflikter
Dr. Gabriel Herrero-Beaumont har modtaget forskningstilskud fra Bristol-Myers-Squibb. Dr. Santos Castañeda har modtaget uddannelses- og forskningstilskud fra Abbott, MSD og Pfizer.