den vigtigste funktionelle bestanddel af den røde blodcelle, der fungerer som det ilttransporterende protein; det er en type hæmoprotein, hvor hvert molekyle er en tetramer, der består af fire monomerer, der holdes sammen af svage bindinger. Det består af to par polypeptidkæder, globinerne, som hver har et tilknyttet hæmmolekyle, der består af jern og et protoporphyrinmolekyle. Symbol Hb.
Jernatomet har en fri valens og kan binde et oxygenmolekyle. Hvert hæmoglobinmolekyle kan således binde ét iltmolekyle. Bindingen af ilt i en monomer øger affiniteten for ilt hos de andre monomerer i tetrameren. Dette gør hæmoglobin til et mere effektivt transportprotein end et monomerisk protein som f.eks. myoglobin.
Syreholdigt hæmoglobin (oxyhæmoglobin) er lysende rødt i farven; hæmoglobin, der ikke er bundet til ilt (deoxyhæmoglobin), er mørkere. Dette forklarer den lyse røde farve i arterielt blod, hvor hæmoglobinet er ca. 97 % mættet med ilt. Veneblod er mørkere, fordi det kun er mættet med ca. 20-70 %, afhængigt af hvor meget ilt vævene bruger. Hæmoglobinets affinitet for kulilte er 210 gange så stærk som dets affinitet for ilt. Det dannede kompleks (carboxyhæmoglobin) kan ikke transportere ilt. Kulilteforgiftning resulterer således i hypoxi og kvælning.
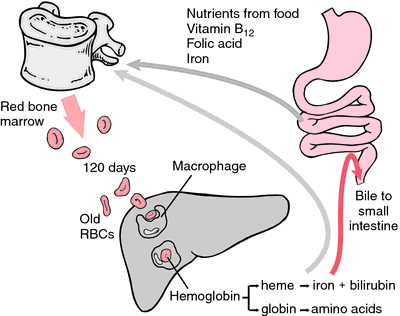
En anden form for hæmoglobin, der ikke kan transportere ilt, er methæmoglobin, hvor jernatomet er oxideret til oxidationstilstand +3. I løbet af de 120 dages levetid for en rød blodcelle oxideres hæmoglobin langsomt til methæmoglobin. Mindst fire forskellige enzymsystemer kan omdanne methæmoglobin tilbage til hæmoglobin. Når disse er defekte eller overbelastede, kan der opstå methæmoglobinæmi, hvor høje methæmoglobinniveauer forårsager dyspnø og cyanose.
En sekundær funktion for hæmoglobin er som en del af blodbuffersystemet. Histidinresterne i globinkæderne fungerer som svage baser for at minimere ændringen i blodets pH-værdi, der opstår, når ilt absorberes og kuldioxid frigives i lungerne, og når ilt leveres og kuldioxid optages fra vævene.
Når erytrocytterne slides eller beskadiges, optages de af makrofager i det reticuloendotheliale system. Hæmets porfyrinring omdannes til galdepigmentet bilirubin, som udskilles af leveren. Jernet transporteres til knoglemarven for at blive inkorporeret i hæmoglobinet i de nyligt dannede erytrocytter.
Hæmoglobinkoncentrationen i blodet varierer med hæmatokriten. De normale værdier for blodets hæmoglobinkoncentration er 13,5 til 18,0 g/100 ml hos mænd og 12,0 til 16,0 g/100 ml hos kvinder. Den normale gennemsnitlige korpuskulære hæmoglobinkoncentration, som er koncentrationen i de røde blodlegemer, er 32 til 36 g/100 ml.
Der er blevet opdaget mange unormale hæmoglobiner, der skyldes mutationer. Nogle har ændret iltaffinitet, nogle er ustabile, og i nogle er jernatomet oxideret, hvilket resulterer i medfødt methæmoglobinæmi. Nogle mutationer resulterer i en nedsat hæmoglobinsyntesehastighed. Alle sådanne tilstande er kendt som hæmoglobinopatier.
Den mest almindelige hæmoglobinopati er seglcellesygdom, som skyldes en mutation, der erstatter den sjette aminosyre i β-kæden, normalt glutaminsyre, med valin. Varianten hæmoglobin α2βS2 er kendt som Hb S. Mutationer, der resulterer i nedsat syntese af en af kæderne, kaldes thalassemias. De kan skyldes deletion af genet for en kæde eller en mutation i det regulatoriske gen, der styrer syntesen af kæden.