Einführung
Rheumatoide Arthritis (RA) ist eine polygene Autoimmunerkrankung, die durch Polyarthritis mit systemischen Manifestationen und erhöhter und schwerer Morbidität gekennzeichnet ist.1,2 Die RA betrifft 0,5-1 % der Bevölkerung und verursacht eine Beeinträchtigung der Lebensqualität, erhebliche körperliche Behinderungen und beträchtliche wirtschaftliche Kosten.3-6 Die klinische Ausprägung der Krankheit ist vielfältig und reicht von milden, selbstlimitierenden Formen bis hin zu einer sehr aggressiven, raschen Entwicklung, die in der Zerstörung des betroffenen Gelenks und der daraus resultierenden Behinderung gipfelt.7
Genetische Studien haben das Vorhandensein eines genetischen Substrats bestätigt, das zum Teil mit bestimmten Genen zusammenhängt, die für Proteine kodieren, die an der Reaktion der T-Zellen beteiligt sind.1 Diese Ergebnisse unterstreichen die Bedeutung der Rolle, die den T-Zellen bei der Auslösung und Aufrechterhaltung der abnormalen Immunantwort bei dieser Krankheit zugeschrieben wird.8
Die Pathogenese der RA ist komplex und betrifft verschiedene Zellpopulationen, die mit der angeborenen und adaptiven Immunantwort zusammenhängen. In der Synovia ansässige Zellen wie fibroblastische Synoviozyten B oder Makrophagen der Intima sowie Entzündungszellen aus dem Blut wie T-Lymphozyten, B-Lymphozyten und Monozyten9 sind an der Pathogenese beteiligt. Sie alle tragen zur aggressiven Umwandlung des Synoviozyten-B-Phänotyps und zur Entwicklung eines intensiven entzündlichen Infiltrats bei, dessen Endergebnis die Zerstörung von Knorpel und subchondralem Knochen ist10,11 (Abb. 1).
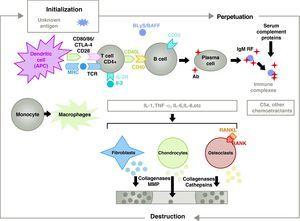
Pathophysiologie der rheumatoiden Arthritis. Allgemeine pathophysiologische Organisation der rheumatoiden Arthritis. AC, Antikörper; BAFF, B-Zell-aktivierender Faktor; BLyS, B-Lymphozyten-Stimulator; CD, Cluster of Differentiation; CPA, Antigen-präsentierende Zelle; CPH, MHC; CTLA4, Lymphozyten-assoziiertes Antigen 4 T zytotoxische C5a-Fraktion Komplement 5a, FR, Rheumafaktor; Ig, Immunglobulin; IL, Interleukin; MMP, Matrix-Metalloproteinasen; RANK, Rezeptor-Aktivator des Nuklearfaktors B kappa; RANKL, Rezeptor-Aktivator-Ligand für den Nuklearfaktor B kappa; RCT, T-Zell-Rezeptor; TNF, Tumor-Nekrose-Faktor.
Die derzeitige Behandlung der RA basiert auf der Verabreichung von krankheitsmodifizierenden Antirheumatika (DMARDs), die allein oder in Kombination eingesetzt werden.12 Diese Medikamente verlangsamen die Gelenkzerstörung, d.h. sie sind in der Lage, den natürlichen Krankheitsverlauf zu modifizieren.4,13 Der Prozentsatz der Patienten mit einem zufriedenstellenden klinischen Ansprechen ist jedoch gering und erfordert bei einem hohen Prozentsatz der Patienten die zusätzliche Verabreichung eines biologischen Arzneimittels.9,13-15
In den letzten Jahren wurden neue Moleküle und therapeutische Zielmoleküle identifiziert, deren Blockade die chronische Entzündungsreaktion verringern oder beseitigen könnte. Eines dieser neuen Moleküle ist Abatacept. Abatacept ist ein vollständig humanisiertes Proteinkonstrukt, das aus der extrazellulären Domäne des humanen zytotoxischen T-Lymphozyten-assoziierten Antigens 4 (CTL4) und einem gentechnisch veränderten Fragment der Fc-Region des humanen Immunglobulins G1 (IgG1) besteht, das die T-Zellen zur Kostenstimulation hemmt, die auf den eigentlichen Kern der Immunreaktion und damit auf den Beginn der Krankheit einwirken.
T-Zellaktivierung
Eine wirksame Immunaktivierung von T-Zellen erfordert die Beteiligung von zwei Gruppen von Membranrezeptoren auf antigenpräsentierenden Zellen (APC)14 (Abb. 1 und 2). Die erste Gruppe ist das Vehikel, mit dem die APCs das zuvor verarbeitete spezifische Antigen an die T-Zelle weiterleiten. Trotz enormer Forschungsanstrengungen ist es noch nicht gelungen, die arthritogenen Antigene zu identifizieren, die die RA auslösen.8 Die Präsentation eines Antigens, gegen das eine spezifische Immunantwort erfolgt, durch APCs wird durch einen trimolekularen Komplex organisiert, der Folgendes umfasst: Moleküle des Haupthistokompatibilitätskomplexes (MHC), die im APC vorhanden sind, das Antigen, gegen das sich die Immunantwort entwickelt, und einen Membranrezeptor auf der T-Zelle (TCR), der für dieses Antigen spezifisch ist15 (Signal- oder Signalweg der Immunantwort 1).
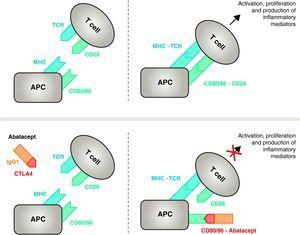
Wirkungsmechanismus von Abatacept. Das Abatacept-Fragment, das die extrazelluläre Domäne von CTLA4 umfasst, bindet an CD80/CD86-Rezeptoren und verhindert oder verdrängt deren Interaktion mit dem CD28-Rezeptor. Auf diese Weise blockiert es selektiv die spezifische Bindung von CD80/CD86 an den CD28-Rezeptor, was pathophysiologisch eine Blockade des zweiten Signals für die Immunaktivierung und damit der Aktivierung von T-Zellen darstellt CPA, antigenpräsentierende Zelle; MHC, Haupthistokompatibilitätskomplex; TCR, T-Zell-Rezeptor.
Um eine vollständige Aktivierung zu verhindern, benötigen T-Zellen einen zweiten Satz von interzellulären Kommunikationsrezeptoren zwischen APCs und T-Zellen, der über kostimulatorische Wege erfolgt und die sogenannte 2-Signal-Immunantwort darstellt.14 Obwohl es mehrere kostimulatorische Wege gibt, ist einer von ihnen wesentlich, nämlich die Bindung der Rezeptoren CD80 (B7-1)/CD86 (B7-2) auf der Membran der CPA mit dem CD28-Rezeptor auf den T-Zellen.10,16 Die gleichzeitige Aktivierung beider löst in den T-Zellen eine intensive intrazelluläre Signalübertragung aus, die für die vollständige Aktivierung, die Proliferation, das Überleben und die Zytokinproduktion unerlässlich ist. 24-48 Stunden nach der Aktivierung der T-Lymphozyten wird durch dieselbe intrazelluläre Signalübertragung ein Regulierungsmechanismus in Gang gesetzt, der darauf abzielt, die Reaktion selbst zu deaktivieren. Dies induziert die Expression von CTLA411 auf der Zellmembran der Lymphozyten, dessen Aufgabe es ist, mit CD28 zu konkurrieren, da es eine größere Bindungsaffinität zu CD80/CD86 hat.17,18
Die Aktivierung beider T-Zell-Untergruppen, CD4+ und CD8+, hängt vom kostimulatorischen Rezeptor CD28 ab. CD4+ T-Zellen sind T-Helferzellen. Sie erkennen die Peptide, die von MHC-Klasse-II-Molekülen auf den APC präsentiert werden. Diese Antigene stammen aus dem exogenen Signalweg, der Krankheitserreger wie Bakterien verarbeitet. Viele Autoimmunkrankheiten sind mit einer pathologischen Reaktion von CD4+ T-Zellen verbunden. Die CD8+ T-Zellen ihrerseits sind zytotoxische Lymphozyten (CTL). CD8+ T-Zellen erkennen Antigene, vor allem virale und Tumorantigene, die von MHC-Molekülen der Klasse I präsentiert werden. Nach der Aktivierung vermitteln CD8+ Zellen die Zerstörung der Zielzellen durch die Produktion von Perforin, Granzymen und Interferon (IFN)-g. Beide Subtypen von T-Zellen werden durch Kostenstimulation mit CD2815 aktiviert, obwohl die Aktivierung von CD8+ T-Zellen weniger von diesem Kostenstimulationsweg abhängig ist. Während nämlich alle CD4+-Zellen CD28 auf ihrer Membran exprimieren, ist dies nur bei etwa 50 % der CD8+-Zellen der Fall.19 Darüber hinaus hat sich gezeigt, dass CD4+-Zellen stärker auf die Bindung von CD2820 reagieren. Außerdem ist der CD28-Promotor keine absolute Voraussetzung für die Aktivierung von CTL.21 All dies würde in der klinischen Praxis einen doppelten therapeutischen Nutzen bringen. Zum einen wirkt Abatacept bevorzugt auf die Zielzelle in der Pathogenese der Krankheit. Zum anderen würde die reduzierte Wirkung auf die Aktivität der CD8+-Lymphozyten ein besseres Sicherheitsprofil in Bezug auf virale und tumorale Komplikationen gewährleisten.
Die Aktivierung von CD4+ T-Zellen ist der Ausgangspunkt für eine proinflammatorische Kaskade mit der Produktion großer Mengen von Zytokinen und der Zellproliferation, die, wenn sie wie bei der RA aufrechterhalten wird, zu einer sehr aktiven chronischen Entzündung führt, die in der Lage ist, die Gewebe zu zerstören, in denen sie ausgelöst wird, im Falle der RA8 meist die Gelenke (Abb. 1). Die Synovialis beginnt zu proliferieren, da Zellen aus dem Blut eindringen, darunter T-Lymphozyten und ihre Subtypen sowie B-Lymphozyten. Die Monozyten differenzieren sich zu Makrophagen und Osteoklasten und aktivieren auch Gelenkknorpelzellen. In diesem Umfeld werden große Mengen an proinflammatorischen Zytokinen wie Interleukin (IL)-1, IL-6 und Tumor-Nekrose-Faktor (TNF) und viele andere gebildet. B-Zellen produzieren auch Autoantikörper wie den Rheumafaktor oder Antikörper gegen citrullinierte Peptide. All dies führt zur Zerstörung nicht nur der Synovialmembran, sondern auch des darunter liegenden Knochens und Knorpels.22
Biotechnologie bei der Behandlung der rheumatoiden Arthritis
Aufgrund der oben genannten Forschungen wurde die biotechnologische Herstellung verschiedener Moleküle zur Blockierung spezifischer Ziele entwickelt und vermarktet. Die erste Generation war durch das Auftreten von TNF-neutralisierenden Medikamenten gekennzeichnet: Etanercept, Infliximab und Adalimumab sowie Anakinra, die die Wirkung von IL-1 hemmen. In der Folge kamen neue Moleküle auf den Markt, wie Abatacept zur Modulation der Kostenstimulation der Immunantwort, Certolizumab und Golimumab zur Blockierung von TNF, Rituximab gegen den CD20-Rezeptor der B-Lymphozyten und Tocilizumab, das IL-6 blockiert.7,23-26
Trotz des großen Sprungs in der therapeutischen Wirksamkeit durch die Einführung dieser Medikamente spricht ein erheblicher Prozentsatz der Patienten, schätzungsweise 25 bis 40 %, nicht auf die derzeit auf dem Markt befindlichen Medikamente oder Biologika an oder ist vom Auftreten unerwünschter Ereignisse betroffen.27-32 Die Notwendigkeit, diese Situation zu verbessern, bleibt ein Ansporn für die Verfolgung und Entwicklung neuer Moleküle, die auf die Regulierung verschiedener therapeutischer Ziele abzielen und die therapeutische Wirksamkeit verbessern könnten, wie im Fall von Abatacept, das selektiv die Aktivierung von T-Zellen moduliert.33
Abatacept ist ein Proteinkonstrukt, das durch rekombinante DNA-Technologie in Hamster-Ovarialzellen hergestellt wird.34,35 Dieses Molekül wurde entwickelt, um in die Regulierung der kostimulatorischen Wege in T-Zellen einzugreifen, die eine wichtige Rolle bei der Pathogenese verschiedener Autoimmunerkrankungen, Infektionen, der Abstoßung transplantierter Organe und der Tumorimmunität spielen.36
Abatacept wird in Kombination mit Methotrexat bei RA-Patienten eingesetzt, die unzureichend auf andere DMARDs, einschließlich Methotrexat (MTX) oder einen TNF-alpha-Hemmer, angesprochen haben oder diese nicht vertragen. Bei polyartikulärer juveniler idiopathischer Arthritis ist es bei Patienten ab 6 Jahren indiziert, die unzureichend auf andere DMARDs einschließlich mindestens eines TNF-neutralisierenden Arzneimittels angesprochen haben.35
Wirkmechanismus von Abatacept
Abatacept ist ein selektiver Modulator des kostimulatorischen CD80/86-CD28-Signals, das, wie bereits erwähnt, für die Aktivierung von T-Zellen unerlässlich ist. Abatacept hemmt die Aktivierung von T-Zellen, indem es selektiv die spezifische Bindung des CD80/CD86-Rezeptors in APC an CD28 auf T-Zellen blockiert (Abb. 2). 22,37 Die pharmakologische Strategie zielt darauf ab, die für die Krankheit charakteristische beschleunigte Immun-/Entzündungsreaktion zu hemmen und die normale Homöostase des Immunsystems wiederherzustellen. Tatsächlich ist der Wettbewerb zwischen endogenem CD28 und CTLA4 um die Bindung an CD80/86 der physiologische Mechanismus, der zur Regulierung und gegebenenfalls zum Abschluss einer normalen Immunantwort eingesetzt wird. Indem Abatacept die Bindung von CD80/86 an CD28 blockiert, hemmt es die Übertragung eines zweiten Signals der Immunantwort, das indirekt ein negatives Signal für die T-Zell-Aktivierung erzeugt. Darüber hinaus hat Abatacept wahrscheinlich eine größere Wirkung bei der Verhinderung der Bildung eines kostimulatorischen Signals in T-Zellen, indem es die bereits aktiven T-Zellen inaktiviert, die nicht an CTLA4
Supporting Drug for Use
1 gebunden sind. Warum gehört Abatacept zur Gruppe der immunmodulatorischen Arzneimittel? Grundsätzlich, weil es zu einer Verarmung der Zellen, insbesondere der T-Zellen, führt, weil es pharmakologisch wirkt, indem es nicht selektiv ein bestimmtes Zytokin blockiert, sondern die radikale Unterdrückung wesentlicher Signalwege für das ordnungsgemäße Funktionieren der Immunantwort verhindert.8
2. Wie verhindert es die Bindung der Fc-Region des Moleküls an seinen Rezeptor? Die Fc-Region von Abatacept ist gentechnisch so verändert, dass sie nicht an die CD16- und CD32-Rezeptoren und nur sehr schwach an den CD64-Rezeptor bindet. Durch diese Konstruktion werden die vom Fc-Rezeptor vermittelten zellulären Reaktionen wie die antikörperabhängige zelluläre Zytotoxizität (ADCC) und die komplementabhängige Zytotoxizität (CDC) umgangen.18 Beide sind mit der Zelllyse verbunden, was bei prolongierten38 Behandlungen zu unerwünschten Wirkungen führen kann. Daher scheint das modifizierte Fragment von IgG1 aktiv zu sein und dadurch unerwünschte Ereignisse aufgrund der ADCC zu verhindern.39
3. Entzündungshemmende Wirkung von Abatacept. Abatacept reduziert viele der Entzündungsmediatoren bei Patienten mit RA signifikant und normalisiert sie, was in mehreren klinischen Studien, die bei der Erforschung des Medikaments verwendet wurden, nachgewiesen wurde.
In einer einjährigen, placebokontrollierten Phase-II-b-Studie bei Patienten mit RA und unzureichendem Ansprechen auf MTX wurden Proben entnommen und die Serumspiegel ausgewählter Marker in den Tagen vor der Infusion gemessen, um die Wirkung von Abatacept auf Mediatoren und proinflammatorische Zytokine zu untersuchen. Eine Gruppe von Patienten erhielt MTX und Abatacept 10 mg/kg nach dem üblichen Schema. Die Kontrollgruppe hingegen wurde mit MTX und Placebo behandelt. Ein Jahr nach der Behandlung hatten sich die Marker in der Abatacept-Gruppe mit 10 mg/kg normalisiert, während sie in der Placebo-Gruppe erhöht blieben (TNF: 7,4 vs. 10,3pg/ml; FR: 159 vs. 225U/l, sIL-2R: vs. 1228,3. 1697,1pg/ml IL-6: 7,3 vs 19,9pg/ml).40
4. Immunogenität. Den Daten über das Medikament zufolge entwickelten nur 187 von 3877 (4,8 %) Patienten mit RA, die bis zu 8 Jahre lang mit Abatacept behandelt wurden, während der Behandlung Antikörper gegen das Medikament.41 Antikörper gegen Abatacept wurden bei Patienten nach Absetzen des Medikaments (>42 Tage nach der letzten Dosis) untersucht, und bei 103 von 1888 (5,5 %) waren sie seropositiv. Im Gegensatz dazu wurden in einer anderen Studie mit 2000 Patienten Abatacept-Antikörper gemessen, und es wurde festgestellt, dass Abatacept eine geringe Immunogenität aufweist.42,43
5. Abatacept und Tuberkulose. TNF ist an der Entzündungsreaktion und Immunpathologie der Tuberkulose (TB) beteiligt. In-vitro-Studien zeigen, dass TNF die phagozytische Aktivität und die mykobakterientötenden Makrophagen erhöht, während sie in vivo an der anfänglichen Bildung und der anschließenden Aufrechterhaltung von Granulomen beteiligt sind, was das Wachstum von Mykobakterien kontrolliert und ihre Ausbreitung begrenzt. In einem chronischen Modell der Reaktivierung latenter Tuberkulose bei Mäusen untersuchten wir die Entwicklung der Infektion bei Mäusen, die mit Abatacept behandelt wurden, im Vergleich zu einer anderen Gruppe, die mit einem monoklonalen Anti-TNF-Mausmedikament behandelt wurde.42 Vier Monate nach der Infektion von C57BL/6-Mäusen mit Mycobacterium tuberculosis und der Bestätigung, dass sie eine latente Tuberkuloseinfektion hatten, wurden die Mäuse 16 Wochen lang mit einer der beiden experimentellen Interventionen behandelt. Nach dieser Zeit starben alle mit Anti-TNF behandelten Mäuse an disseminierter TB mit einer durchschnittlichen Überlebenszeit von 44 Tagen. Im Gegensatz dazu starb keine der mit Abatacept behandelten Mäuse.
Während sich die Konzentration von IFN-g im Serum in der Abatacept-Gruppe nicht veränderte, war sie bei Mäusen mit Anti-TNF erhöht. Dieser Anstieg wurde auf die verstärkte Infiltration von CD4+ und CD8+ zurückgeführt, die durch die weite Ausbreitung der Bakterienkolonien verursacht wurde.
Während also Mäuse, die mit Anti-TNF behandelt wurden, eine 100%ige Sterblichkeit aufwiesen, veränderte Abatacept nicht die Fähigkeit der Mäuse, eine Entzündungsreaktion zu organisieren, die die Ausbreitung der Tuberkulose kontrollieren kann. Allerdings gibt es noch nicht genügend klinische Daten, um diese Ergebnisse beim Menschen zu bestätigen.
6. Antiresorptive Wirkung von Abatacept auf den Knochenumbau. Die Osteoklastenaktivität ist bei RA erhöht, und zwar sowohl im Gelenk, was zu Knochenerosionen führt, als auch systemisch, wobei ein Niveau erreicht wird, das mit einer generalisierten Osteoporose assoziiert ist.44,45
In der Tat wurde ein Anstieg des Liganden Rezeptor-Aktivator des Nuklearfaktors NF-kB (RANKL) in der Synovialmembran nachgewiesen.45,46 Abatacept hemmt dosisabhängig die Osteoklastenbildung bei Mäusen und die in vitro ermittelte osteoklastogene Aktivität. Dies wurde an Mäuse-Osteoklasten untersucht, die auf Dentinplatten kultiviert wurden, wobei die Anzahl der Resorptionsgruben nach 6 Tagen der Zugabe verschiedener Abatacept-Dosen gemessen wurde.47
Das Medikament verringerte die Fläche der Knochenresorption erheblich. Diese Daten deuten darauf hin, dass Abatacept ein Molekül ist, das direkt an die Vorläuferzellen der Osteoklasten bindet und deren Differenzierung hemmt. Dieser Mechanismus könnte die anti-erosive Wirkung des Medikaments bei Patienten mit RA erklären. Tatsächlich zeigten Patienten, die mit Abatacept behandelt wurden, einen rückläufigen Trend bei den RANK- und RANKL-Ligandenspiegeln in der Synovialis, die alle mit einem Anstieg des Osteoprotegerins einhergingen.48 Obwohl der genaue Mechanismus, der dieser Beobachtung zugrunde liegt, unklar ist, korrelieren diese Ergebnisse gut mit der radiologischen Verbesserung, die bei den mit Abatacept behandelten Patienten beobachtet wurde.
7. Auswirkungen von Abatacept auf andere Immunzellen. Obwohl die APC die Zielzelle ist, die Abatacept bindet, und Makrophagen ebenfalls CD80/86-Rezeptoren auf ihrer Oberfläche exprimieren, gibt es nur wenige Studien, die die Wirkung des Arzneimittels auf die Aktivität dieser Zellen untersuchen. Eine kürzlich durchgeführte In-vitro-Studie hat gezeigt, dass Makrophagen eine ausgeprägte CD80/86-Rezeptorexpression aufweisen und die Behandlung mit Abatacept die Produktion von Zytokinen49 erheblich reduziert. Diese Ergebnisse deuten darauf hin, dass der Wirkmechanismus des Medikaments auf die Regulierung der Makrophagenabstammung ausgedehnt werden könnte, die eine Schlüsselrolle in der Pathogenese der Krankheit spielt.
Abatacept unterdrückt auch die follikuläre Migration antigenspezifischer T-Zellen und folglich die Zusammenarbeit zwischen T-Zellen und follikulären B-Zellen im Lymphknoten. Dieser Befund wurde in situ in Lymphknoten von BALB/c50-Mäusen beobachtet. Nach der Transfusion solcher Mäuse mit antigenspezifischen, vorstimulierten T-Zellen zeigte sich bei einer anschließenden Immunisierung von Mäusen eine Vermehrung der T-Zellen und eine Migration in den B-Lymphozyten-Bereich. Bei Mäusen, die mit Abatacept behandelt wurden, wurde die Proliferation und Migration der T-Zellen blockiert, so dass sie in den meisten Fällen nur noch im Parakortex der Lymphknoten vorkamen. Somit reduziert eine längere Behandlung mit Abatacept die Proliferation, Mobilität und Verteilung der intraganglionären Autoantigen-Gedächtnis-Lymphozyten, was zu einem Rückgang der Autoantikörper führen könnte.
Schlussfolgerungen zum Wirkmechanismus von Abatacept
Abatacept ist ein vollständig humanisiertes Proteinkonstrukt, das aus der extrazellulären Domäne des menschlichen zytotoxischen T-Lymphozyten-assoziierten Antigens 4 (CTL4) und einem genetisch veränderten Fragment der Fc-Region von IgG1 besteht, Das Medikament hemmt die Aktivierung von T-Zellen durch selektive Blockierung der spezifischen Bindung von CD80/CD86 an den CD28-Rezeptor und hemmt somit die T-Zell-Proliferation und die Immunreaktion von B-Lymphozyten Diese pharmakologische Wirkung führt zu einer Verringerung der Entzündungsmediatoren bei Patienten mit RA und zu einer sicheren und wirksamen klinischen Reaktion.
Interessenkonflikt
Dr. Gabriel Herrero-Beaumont hat Forschungsgelder von Bristol-Myers-Squibb erhalten. Dr. Santos Castañeda hat Ausbildungs- und Forschungszuschüsse von Abbott, MSD und Pfizer erhalten.