Editorial zum Forschungsthema
The Role of AAA+ Proteins in Proteinreparatur und -abbau
ATPasen, die mit verschiedenen zellulären Aktivitäten assoziiert sind (AAA+), umfassen eine Superfamilie von Proteinen, die eine Vielzahl von für die Zellphysiologie wichtigen Funktionen ausüben, Dazu gehören die Kontrolle der Proteinhomöostase, der DNA-Replikation, der Rekombination, des Chromatinumbaus, der Verarbeitung ribosomaler RNA, des molekularen Targetings, der Organellenbiogenese und der Membranfusion (Hanson und Whiteheart, 2005; Erzberger und Berger, 2006; Snider et al., 2008). Mitglieder dieser Superfamilie sind durch das Vorhandensein der so genannten AAA+-Domäne definiert, die die kanonischen Walker A- und B-Motive enthält, die für die ATP-Bindung und -Hydrolyse erforderlich sind (Hanson und Whiteheart, 2005). In der Regel kodieren Genome für etwa zehn bis mehrere hundert Mitglieder der AAA+-Familie (Tabelle 1; Finn et al., 2017), von denen angenommen wird, dass sie jeweils an spezifische funktionelle Nischen angepasst sind, die präzise Mechanismen der Substraterkennung und -verarbeitung erfordern (Hanson und Whiteheart, 2005). Die auffällige adaptive Ausstrahlung von AAA+-Proteinen, die in verschiedenen Umgebungen wirken, veranschaulicht den vielseitigen Nutzen der AAA+-Domäne (Erzberger und Berger, 2006). AAA+-Proteine bilden in der Regel hexamere Komplexe und fungieren als Motoren für den Umbau anderer Proteine, DNA/RNA oder Multikomponentenkomplexe (Abbildung 1). Viele Chaperone und ATP-abhängige Proteasen sind oder haben Untereinheiten, die zu dieser Superfamilie gehören (Abbildung 1; Olivares et al., 2016).
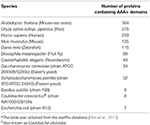
Table 1. Anzahl der AAA+-Proteine in Modellorganismena.
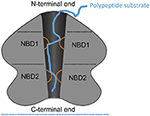
Abbildung 1. Schematische Darstellung von ClpA als Beispiel für ein AAA+-Hexamer. Schematische Darstellung der Domänenarchitektur und Interaktionen von ClpA als Beispiel für ein AAA+-Hexamer mit zwei AAA+-Domänen pro Monomer. Hier wird eine Seitenansicht des ClpA-Hexamers gezeigt, um den zentralen, polypeptidleitenden Kanal darzustellen. ClpA enthält drei Domänen, darunter eine N-terminale Domäne und zwei AAA+-Domänen: Nukleotid-Bindungsdomäne 1 und 2 (NBD1 und NBD2). Die N-terminale Domäne interagiert mit Regulatoren der Substratspezifität, während das C-terminale Ende mit der gekammerten Protease ClpP interagiert. Die beweglichen Schleifen von NBD1 und NBD2 (orange) ragen in den zentralen Kanal hinein und interagieren mit dem Polypeptidsubstrat (blau), wodurch die Kopplung von ATP-Hydrolyse und Polypeptid-Translokation durch den zentralen Kanal ermöglicht wird.
In den letzten Jahren wurden erhebliche Fortschritte bei der Identifizierung der Struktur und des Funktionsmechanismus einer großen Anzahl von AAA+-Proteinen erzielt (Gates et al., 2017; Puchades et al., 2017; Ripstein et al., 2017; Zehr et al., 2017). In diesem Forschungsthema werden mehrere Elemente dieses spannenden Fortschritts in 21 Artikeln vermittelt, die eine detaillierte strukturelle und mechanistische Sicht auf mehrere AAA+-Chaperone und Proteasen umfassen, darunter: ClpX (Alhuwaider und Dougan; Bittner et al.; Elsholz et al.; LaBreck et al.; Vass et al.), ClpA (Bittner et al.; Duran et al.), ClpB und Hsp104 (Chang et al.; Duran et al.; Franke et al.; Johnston et al.), Hsp78 (Abrahão et al.), ClpC (Alhuwaider und Dougan; Elsholz et al.), ClpE (Elsholz et al.), Pontin (Mao und Houry), Reptin (Mao und Houry), FtsH (Alhuwaider und Dougan), 19S Proteasom (Snoberger et al.; Yedidi et al.), Lon (Alhuwaider und Dougan; Bittner et al.; Fishovitz et al.), p97 (Hänzelmann und Schindelin; Saffert et al.; Ye et al.), Pex1/6 (Saffert et al.), CbbQ (Mueller-Cajar), Rubisco-Aktivase (Bhat et al.), Torsine (Chase et al.) und mitochondriale AAA+ Proteasen (Glynn). Hier stellen wir diese faszinierenden Arbeiten vor.
Studien über ClpXP, Lon und verwandte ATP-abhängige Proteasen
In ihrem Forschungsartikel „The Protein Chaperone ClpX Targets Native and Non-native Aggregated Substrates for Remodeling, Disassembly, and Degradation with ClpP“ (Das Protein-Chaperon ClpX zielt auf native und nicht-native aggregierte Substrate für Umbau, Abbau und Degradation mit ClpP), führen LaBreck et al. führen eine Reihe eleganter Experimente durch, um nachzuweisen, dass ClpX eine Disaggregase-Aktivität gegenüber Polypeptiden besitzt, die spezifische ClpX-Erkennungssignale enthalten (LaBreck et al.). In Gegenwart von ClpP koppelt ClpX die Disaggregation dieser Substrate an deren Abbau. Wichtig ist auch der Nachweis, dass ClpXP die Anhäufung von Aggregaten verhindert, die von Proteinen mit ClpX-Erkennungssignalen in vivo gebildet werden (LaBreck et al.). Diese Studien beleuchten ClpX als eine Protein-Disaggregase, die bisher unterschätzt wurde.
In ihrem Forschungsartikel „The Essential Role of ClpXP in Caulobacter crescentus Requires Species Constrained Substrate Specificity“ (Die wesentliche Rolle von ClpXP in Caulobacter crescentus erfordert speziesbeschränkte Substratspezifität) untersuchen Vass et al. die speziesspezifischen Funktionen von ClpX (Vass et al.). Interessanterweise ist ClpX in einigen Arten wie C. crescentus essentiell, in anderen Bakterien wie E. coli jedoch nicht (Vass et al.). Interessanterweise war E. coli ClpX nicht in der Lage, C. crescentus ClpX in vivo zu ergänzen (Vass et al.). Diese fehlende Aktivität war auf artspezifische Unterschiede in der N-terminalen Domäne von ClpX zurückzuführen, die für die Verarbeitung der Replikationsklammer-Lader-Untereinheit DnaX in C. crescentus entscheidend ist. Daher können kleine Unterschiede in der ClpX-Spezifität für bestimmte Bakterienarten besonders kritisch sein.
In ihrer Übersichtsarbeit über „Functional Diversity of AAA+ Protease Complexes in Bacillus subtilis“ diskutieren Elsholz et al. die Funktionen mehrerer AAA+ Proteasen in B. subtilis, nämlich: ClpCP, ClpEP, ClpXP, ClpYQ, LonA/B, und FtsH (Elsholz et al.). Sie erörtern, wie verschiedene Stressreaktionen ihre Expression kontrollieren und welche Phänotypen bei der Deletion dieser verschiedenen Proteasen beobachtet werden. Die Fähigkeit einiger dieser Proteasen, Kompetenz, Sporulation, Motilität und Biofilmbildung zu kontrollieren, wird beschrieben. Abschließend erörtern die Autoren, wie diese Proteasen zur Entwicklung neuer Antibiotika eingesetzt werden können.
In ihrer Übersichtsarbeit mit dem Titel „AAA+ Machines of Protein Destruction in Mycobacteria“ (AAA+ Machines of Protein Destruction in Mycobacteria) erörtern Alhuwaider et al. (Alhuwaider und Dougan) die jüngsten Fortschritte bei der Bestimmung der Struktur und Funktion von AAA+ Proteasen von Mykobakterien. Diese Proteasen sind: ClpXP1P2, ClpC1P1P2, Lon, FtsH und Mpa. Die Autoren erörtern auch das in Mykobakterien vorhandene Pup-Proteasom-System (PPS), das dem Ubiquitin-Proteasom-System in Eukaryonten entspricht. Alhuwaider et al. schließen mit einer Diskussion über neuartige Verbindungen, die die Aktivität von ClpP1P2 dysregulieren oder hemmen, und andere, die ClpC1 dysregulieren. Diese Verbindungen haben vielversprechende Aktivitäten gegen Mykobakterien.
In einem Forschungsartikel mit dem Titel „The Copper Efflux Regulator CueR Is Subject to ATP-Dependent Proteolysis in Escherichia coli“ zeigen Bittner et al., dass die AAA+-Proteasen Lon, ClpXP und ClpAP für den Abbau von E. coli CueR verantwortlich sind, das ein Transkriptionsfaktor ist, der die Induktion des Kupfer-Efflux-Cue-Systems steuert (Bittner et al.). Die Autoren fanden heraus, dass die Erkennung von CueR durch die AAA+-Proteasen den zugänglichen C-Terminus von CueR erfordert. Sie kommen zu dem Schluss, dass ATP-abhängige Proteasen für die Kupferhomöostase in E. coli erforderlich sind.
Fishovitz et al. führen in ihrem Forschungsartikel mit dem Titel „Utilization of Mechanistic Enzymology to Evaluate the Significance of ADP Binding to Human Lon Protease“ (Fishovitz et al.) einen detaillierten Vergleich zwischen menschlichem und E. coli Lon durch. Anhand einer detaillierten mechanistischen Studie fanden sie heraus, dass menschliches Lon im Gegensatz zu E. coli Lon eine geringe Affinität für ADP hat, obwohl es vergleichbare kcat- und KM-Werte bei der ATPase-Aktivität aufweist. Sie schlagen vor, dass menschliches Lon nicht durch einen ADP/ATP-Austauschmechanismus reguliert wird, der durch ein Substrat gefördert wird. Diese Unterschiede zwischen menschlichem und E. coli Lon könnten die künftige Entwicklung speziesspezifischer Lon-Inhibitoren ermöglichen.
In seiner Übersichtsarbeit über „Multifunktionelle mitochondriale AAA-Proteasen“ erörtert Dr. Glynn die beiden mitochondrialen AAA-Proteasen, i-AAA und m-AAA (Glynn). Beide sind mitochondriale Innenmembranproteine. Allerdings projiziert die i-AAA die ATPase- und Proteasedomänen in den mitochondrialen Intermembranraum, während die m-AAA-Protease die katalytischen Domänen in die Matrix projiziert. Die Strukturen dieser Proteasen werden ebenso diskutiert wie ihre Funktionsmechanismen. Die Proteasen können einen vollständigen Substratabbau durchführen, aber auch nur bestimmte Substrate spalten, wie z.B. MrpL32 und Atg32.
ClpB und Hsp104
In ihrem Mini-Review, „Structural Elements Regulating AAA+ Protein Quality Control Machines“, diskutieren Chang et al. wie die Porenschleife 1, das Inter-Subunit Signaling-Motiv und das Pre-Sensor I Insert-Motiv zur Aktivität von zwei Hsp100-Disaggregasen, dem bakteriellen ClpB und dem Hefe-Hsp104, beitragen könnten (Chang et al.). Sie schlagen ein Modell vor, wie diese Strukturelemente die Kopplung des AAA+-ATPase-Zyklus mit der Substrattranslokation durch den zentralen Kanal von ClpB und Hsp104 ermöglichen könnten. Es wird angenommen, dass dieser Prozess der Polypeptid-Translokation die Grundlage dafür bildet, wie ClpB und Hsp104 Polypeptide aus aggregierten Strukturen extrahieren (Chang et al.).
Duran et al. liefern in ihrer Übersichtsarbeit eine „Vergleichende Analyse der Struktur und Funktion der AAA+-Motoren ClpA, ClpB und Hsp104: Gemeinsame Fäden und unterschiedliche Funktionen“ (Duran et al.). Sie diskutieren die Fähigkeit dieser drei AAA+-Proteine (ClpA, ClpB und Hsp104), Polypeptide durch ihre hexameren Komplexe zu verlagern. Alle diese Proteine haben zwei AAA+-Domänen und sind für die Entfaltung von Proteinen bekannt. Wichtig ist, dass ClpB und Hsp104 auch als Disaggregasen fungieren, während ClpA einen Komplex mit der ClpP-Protease bilden kann. Die Autoren betonen die Notwendigkeit, die kinetischen Mechanismen dieser Motorproteine mit kinetischen Methoden im Übergangszustand zu untersuchen. Sie beschreiben, wie sie mit Hilfe solcher Methoden zeigen konnten, dass ClpA beispielsweise Polypeptide mit einer Geschwindigkeit von etwa 20 aa s-1 transloziert, während die Translokationsrate von ClpA im Komplex mit der ClpP-Protease mit etwa 35 aa s-1 noch höher ist. Die Autoren erörtern auch die Bedeutung des Chaperons Hsp70 für die Funktion von ClpB/Hsp104 und die Beobachtung von Spezies-Spezifität in der Interaktion zwischen Hsp70 und ClpB/Hsp104.
In ihrem Forschungsartikel mit dem Titel „Mutant Analysis Reveals Allosteric Regulation of ClpB Disaggregase“ führen Franke et al. eine Mutationsanalyse an der E. coli ClpB Disaggregase durch, um ihre allosterische Regulation zu charakterisieren (Franke et al.). ClpB kann in eine N-terminale Domäne und zwei AAA+-Domänen unterteilt werden, die durch eine helikale Region, die M-Domäne, getrennt sind. Die Autoren identifizieren einen hoch konservierten Rest in der ersten AAA+-Domäne, A328. Bei der ClpB-A328V-Mutante wurde eine sehr hohe ATPase-Aktivität festgestellt, und sie wies eine zelluläre Toxizität auf. Unerwarteterweise war die hohe ATPase-Aktivität von ClpB-A328V hauptsächlich auf den zweiten AAA+-Ring zurückzuführen, wie durch Amid-Hydrogenaustausch-Massenspektrometrie festgestellt wurde. Die Autoren kommen zu dem Schluss, dass A328 ein entscheidender Rest bei der Kontrolle der ATP-Hydrolyse in beiden AAA+-Ringen von ClpB ist.
In ihrem Forschungsartikel mit dem Titel „Substrate Discrimination by ClpB and Hsp104“ beschreiben Johnston et al. die angeborenen Substratpräferenzen von ClpB und Hsp104 in Abwesenheit der Chaperonsysteme DnaK und Hsp70 (Johnston et al.). Sie zeigen, dass die Substratspezifität durch die erste AAA+-Domäne in jedem Protein bestimmt wird. Zu dieser Schlussfolgerung gelangten sie, indem sie die beiden Chaperone auf ihre Fähigkeit testeten, auf verschiedene Modellsubstrate zu wirken. Außerdem testeten sie verschiedene Chimären der beiden Chaperone.
In „Hsp78 (78 kDa Heat Shock Protein), a Representative AAA Family Member Found in the Mitochondrial Matrix of Saccharomyces cerevisiae,“ diskutieren Abrahão et al. die Struktur und Funktion von Hsp78 (Abrahao et al.). Hsp78 ist das mitochondriale Paralogon von Hsp104, das für die Proteindisaggregation und -reaktivierung zuständig ist (Abrahao et al.). Merkwürdigerweise gingen Hsp104 und Hsp78 beim Übergang von Protozoen zu Metazoen verloren (Abrahao et al.). Abrahao et al. diskutieren jedoch die Existenz von ANKCLP, das neben Hsp78 und Hsp104 in Protozoen auftritt und den evolutionären Übergang zu Metazoen überlebt. ANKCLP besitzt eine AAA+-Domäne, die der Nukleotid-bindenden Domäne 2 (NBD2) von Hsp104 und Hsp78 ähnelt, ist aber ansonsten sehr unterschiedlich. Interessanterweise verursachen Mutationen in ANKCLP 3-Methylglutaconsäureurie, progressive Hirnatrophie, geistige Behinderung, kongenitale Neutropenie, Katarakte und Bewegungsstörungen beim Menschen (Abrahao et al.).
p97
In ihrer Übersichtsarbeit „Structure and Function of p97 and Pex1/6 Type II AAA+ Complexes“ (Struktur und Funktion von p97 und Pex1/6 Typ II AAA+ Komplexen) diskutieren Saffert et al. zwei verschiedene AAA+ Komplexe, die ubiquitinierte Substratproteine umbauen (Saffert et al.). Eine Funktion von p97 besteht darin, ubiquitinierte Substrate während des ER-assoziierten Abbaus von der ER-Membran zum Proteasom zu verlagern (Saffert et al.). Im Gegensatz dazu ist Pex1/Pex6 ein heterohexamerer Motor, der abwechselnd aus den Untereinheiten Pex1 und Pex6 besteht und für die Biogenese und Funktion von Peroxisomen wesentlich ist. Jüngste Kryo-Elektronenmikroskopie (Kryo-EM)-Strukturen von p97 und Pex1/6 werden erörtert und wichtige strukturelle Unterschiede hervorgehoben.
In ihrer Übersichtsarbeit mit dem Titel „A Mighty ‚Protein Extractor‘ of the Cell: Structure and Function of the p97/CDC48 ATPase“ fassen Ye et al. das aktuelle Wissen über die Struktur und Funktion von p97 und seine Rolle bei verschiedenen Krankheiten zusammen (Ye et al.). p97 hat zwei AAA+-Domänen, die durch einen kurzen Linker verbunden sind. Außerdem besitzt es eine N-terminale Domäne, die seine Interaktionen mit verschiedenen Adaptorproteinen vermittelt. Die Autoren erörtern ausführlich die Struktur von p97 und die Auswirkungen von Nukleotiden auf seine verschiedenen Konformationen. Diese Studien basieren auf der Anwendung von Techniken wie EM, Röntgenkristallographie und Hochgeschwindigkeits-Rasterkraftmikroskopie. Anschließend erörtern die Autoren die multizellulären Funktionen dieses hochkonservierten Proteins, einschließlich seiner Rolle beim ER-assoziierten Proteinabbau (ERAD), beim mitochondrienassoziierten Abbau (MAD) durch Extraktion von Polypeptiden aus der äußeren Mitochondrienmembran und beim ribosom-assoziierten Abbau (RAD). Schließlich geben Ye et al. einen Überblick über p97-Mutationen, die zu verschiedenen menschlichen Krankheiten führen, wie IBMPFD (Inclusion Body Myopathy associated with Paget’s disease of the bone and Frontotemporal Dementia)], FALS (familiäre amyotrophe Lateralsklerose), CMT2Y (Charcot-Marie-Tooth-Krankheit, Typ 2Y), hereditäre spastische Paraplegien (HSP), Parkinson-Krankheit (PD) und Alzheimer-Krankheit (AD).
In ihrer Übersichtsarbeit über p97 mit dem Titel „The Interplay of Cofactor Interactions and Post-translational Modifications in the Regulation of the AAA+ ATPase p97“ (Das Zusammenspiel von Kofaktorinteraktionen und posttranslationalen Modifikationen bei der Regulation der AAA+ ATPase p97) erörtern Hänzelmann und Schindelin, wie verschiedene Kofaktoren die Aktivität der p97 ATPase modulieren (Hanzelmann und Schindelin). Sie betonen, dass die Fähigkeit von p97, an einer Vielzahl von zellulären Prozessen beteiligt zu sein, auf die große Anzahl von Cofaktoren zurückzuführen ist, die mit diesem Protein interagieren. Sie klären drei verschiedene Klassen von p97-Cofaktoren auf, nämlich: (i) substratbeschaffende Cofaktoren wie UBA-UBX-Proteine und UFD1-NPL4, (ii) substratverarbeitende Cofaktoren wie Ubiquitin-(E3)-Ligasen und Deubiquitinasen (DUBs) und (iii) regulierende Cofaktoren wie die UBX-Proteine, die p97-Hexamere sequestrieren oder recyceln können. Die Autoren erörtern auch die Rolle der posttranslationalen Modifikationen auf die Aktivität von p97 und auf seine Wechselwirkungen mit seinen Cofaktoren und Substraten.
AAA+ Proteine des Proteasoms
In „AAA-ATPases in Protein Degradation,“ Yedidi et al. die Aktivitäten von Rpt1, Rpt2, Rpt3, Rpt4, Rpt5 und Rpt6, den AAA+-ATPasen des eukaryotischen Proteasoms, sowie einiger ihrer bakteriellen Verwandten wie PAN, Mpa und VAT (Yedidi et al.). Sie konzentrieren sich auf neue Technologien, um zu verstehen, wie diese AAA+ ATPasen funktionieren, indem sie ungefaltete Polypeptide in die proteolytische Kammer der Protease verlagern (Yedidi et al.). Konformationsänderungen innerhalb des AAA+-Rings und der angrenzenden Proteasekammer scheinen einen peristaltischen Pumpmechanismus zu erzeugen, um Substrate zum Abbau zu befördern (Yedidi et al.).
In ihrem Forschungsartikel „The Proteasomal ATPases Use a Slow but Highly Processive Strategy to Unfold Proteins“ (Die proteasomalen ATPasen verwenden eine langsame, aber hochgradig prozessive Strategie zur Entfaltung von Proteinen), stellen Snoberger et al. dass proteasomale AAA+-Proteine einen langsamen, aber hoch prozessiven Motormechanismus verwenden, um Substrate in die proteolytische Höhle des Proteasoms zu befördern (Snoberger et al.). Dieser Mechanismus steht im Gegensatz zu ClpX, das einen schnellen, aber weniger prozessiven Motormechanismus einsetzt, um Substrate zum Abbau an die ClpP-Protease zu liefern. Diese Unterschiede in der Motorik könnten sich als Reaktion auf die unterschiedlichen Anforderungen ihrer spezifischen Klientel entwickelt haben.
Rubisco-Aktivasen
In ihrer Übersicht über „Rubisco Activases: AAA+ Chaperones Adapted to Enzyme Repair“ diskutieren Bhat et al. die einzigartige Funktion der Rubisco-Aktivase (Rca) beim Umbau von Rubisco (Bhat et al.). Rca ist ein AAA+-Chaperon, das in photosynthetischen Organismen von Bakterien bis zu höheren Pflanzen hoch konserviert ist. Rubisco ist ein Ribulose-1,5-Bisphosphat-Carboxylase/Oxygenase-Enzym, das während der Photosynthese an der Fixierung von CO2 in der Atmosphäre beteiligt ist. Es ist das am häufigsten vorkommende Protein auf der Erde und das Schlüsselenzym bei der Synthese aller organischen Stoffe auf unserem Planeten. Rubisco ist jedoch ein schwaches Enzym und wird leicht durch Nebenprodukte seiner katalytischen Reaktionen oder durch Verbindungen gehemmt, die von einigen Pflanzen unter schlechten Lichtverhältnissen synthetisiert werden. Rca hat die Aufgabe, Rubisco von solchen problematischen Hemmungen zu befreien oder zu „heilen“. Die Autoren erörtern die Struktur von Rca aus verschiedenen Arten und die möglichen Mechanismen seiner Funktion.
Dr. Mueller-Cajar gibt einen Überblick über „The Diverse AAA+ Machines that Repair Inhibited Rubisco Active Sites“ (Mueller-Cajar). Er erörtert das Vorhandensein von drei evolutionär unterschiedlichen Klassen von Rubisco-Aktivasen (Rcas): (1) grüne und (2) rote Rcas, die hauptsächlich in photosynthetischen Eukaryoten der grünen bzw. roten Plastidenlinie vorkommen, und (3) CbbQO, die in chemoautotrophen Bakterien vorkommt. Er erörtert die Evolution dieser Aktivasen und ihre mögliche Verwendung in der synthetischen Biologie zur Steigerung der Rubisco-Aktivität in Pflanzen.
Torsin
In ihrem perspektivischen Artikel, „Torsin ATPases: Harnessing Dynamic Instability for Function“ diskutieren Chase et al. die Torsins, die phylogenetisch auch mit NBD2 von Hefe Hsp104 verwandt sind (Chase et al.). Torsine sind die einzigen AAA+-ATPasen, die innerhalb des ER und der damit verbundenen Kernhülle lokalisiert sind (Chase et al.). Interessanterweise verursachen Mutationen in TorsinA die DYT1-Dystonie, eine neurologische Störung beim Menschen (Chase et al.). Torsine weisen eine schwache ATPase-Aktivität auf, die durch die Komplementierung der aktiven Stelle aufgrund des Zusammenbaus mit den spezifischen akzessorischen Kofaktoren LAP1 und LULL1 verstärkt wird (Chase et al.). Chase et al. vermuten, dass der dynamische Auf- und Abbau von Torsin/Kofaktor-Komplexen eine wichtige Rolle für ihre Funktion beim Kerntransport und beim Aufbau des Kern-Poren-Komplexes spielt (Chase et al.).
Pontin und Reptin
In ihrer ausführlichen Übersichtsarbeit „The Role of Pontin and Reptin in Cellular Physiology and Cancer Etiology“ (Die Rolle von Pontin und Reptin in der Zellphysiologie und Krebsentstehung) erörtern Mao und Houry die vielfältigen Funktionen der hochkonservierten AAA+ ATPasen Pontin und Reptin (Mao und Houry). Diese beiden Proteine arbeiten in der Regel zusammen als Komplex, können aber auch unabhängig voneinander funktionieren. Die Autoren heben die Rolle von Pontin und Reptin bei der Umstrukturierung von Chromatin hervor. Sie erörtern auch, wie Pontin und Reptin die Transkriptionsaktivitäten mehrerer Proto-Onkogene wie MYC und β-Catenin modulieren. Mao und Houry erläutern, wie Pontin und Reptin für den Aufbau von PIKK-Signalkomplexen sowie für Telomerase, mitotische Spindel, RNA-Polymerase II und snoRNPs erforderlich sind. Die Autoren schließen mit einem Überblick über die derzeitigen Bemühungen zur Identifizierung von Pontin- und Reptin-Inhibitoren, die als neuartige Krebsmedikamente entwickelt werden sollen.
Abschließende Bemerkungen
Zusammenfassend lässt sich sagen, dass diese Sammlung von 21 Artikeln eine Reihe wichtiger struktureller und mechanistischer Aspekte von AAA+-Proteinen beleuchtet, die an der Reparatur und dem Abbau von Proteinen beteiligt sind. Wir sind gespannt, wie sich das Feld im Zuge der laufenden Kryo-EM-Revolution (Egelman, 2016) weiter entwickeln wird. Wir erwarten, dass die Kryo-EM ein tieferes Verständnis dafür ermöglichen wird, wie diese faszinierenden molekularen Maschinen in verschiedenen Situationen funktionieren (Gates et al., 2017; Puchades et al., 2017; Ripstein et al., 2017; Zehr et al, 2017).
Autorenbeiträge
Alle aufgeführten Autoren haben einen substanziellen, direkten und intellektuellen Beitrag zu dieser Arbeit geleistet und sie zur Veröffentlichung freigegeben.
Erklärung zu Interessenkonflikten
Die Autoren erklären, dass die Forschung in Abwesenheit jeglicher kommerzieller oder finanzieller Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Danksagungen
Die Arbeit im Labor von WH auf diesem Gebiet wird durch einen Canadian Institutes of Health Research Project Grant (PJT-148564) finanziert. JS wird vom NIH Grant R01GM099836 unterstützt.
Egelman, E. H. (2016). Die aktuelle Revolution in der Kryo-EM. Biophys. J. 110, 1008-1012. doi: 10.1016/j.bpj.2016.02.001
PubMed Abstract | CrossRef Full Text | Google Scholar
Erzberger, J. P., and Berger, J. M. (2006). Evolutionäre Beziehungen und Strukturmechanismen von AAA+-Proteinen. Annu. Rev. Biophys. Biomol. Struct. 35, 93-114. doi: 10.1146/annurev.biophys.35.040405.101933
PubMed Abstract | CrossRef Full Text | Google Scholar
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro im Jahr 2017 – jenseits von Proteinfamilien- und Domänenannotationen. Nucleic Acids Res. 45, D190-D199. doi: 10.1093/nar/gkw1107
PubMed Abstract | CrossRef Full Text | Google Scholar
Gates, S. N., Yokom, A. L., Lin, J., Jackrel, M. E., Rizo, A. N., Kendsersky, N. M., et al. (2017). Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357, 273-279. doi: 10.1126/science.aan1052
PubMed Abstract | CrossRef Full Text | Google Scholar
Hanson, P. I., and Whiteheart, S. W. (2005). AAA+ proteins: have engine, will work. Nat. Rev. Mol.Cell Biol. 6, 519-529. doi: 10.1038/nrm1684
PubMed Abstract | CrossRef Full Text | Google Scholar
Olivares, A. O., Baker, T. A., and Sauer, R. T. (2016). Mechanistische Einblicke in bakterielle AAA+-Proteasen und Protein-Umbaumaschinen. Nat. Rev. Microbiol. 14, 33-44. doi: 10.1038/nrmicro.2015.4
PubMed Abstract | CrossRef Full Text | Google Scholar
Puchades, C., Rampello, A. J., Shin, M., Giuliano, C. J., Wiseman, R. L., Glynn, S. E., et al. (2017). Struktur der mitochondrialen Innenmembran-AAA+-Protease YME1 gibt Einblick in die Substratverarbeitung. Science 358:eaao0464. doi: 10.1126/science.aao0464
PubMed Abstract | CrossRef Full Text | Google Scholar
Ripstein, Z. A., Huang, R., Augustyniak, R., Kay, L. E., and Rubinstein, J. L. (2017). Struktur einer AAA+ Unfoldase bei der Entfaltung von Substrat. Elife 6:e25754. doi: 10.7554/eLife.25754
PubMed Abstract | CrossRef Full Text | Google Scholar
Snider, J., Thibault, G., and Houry, W. A. (2008). Die AAA+-Superfamilie der funktionell vielfältigen Proteine. Genome Biol. 9:216. doi: 10.1186/gb-2008-9-4-216
PubMed Abstract | CrossRef Full Text | Google Scholar
Zehr, E., Szyk, A., Piszczek, G., Szczesna, E., Zuo, X., and Roll-Mecak, A. (2017). Katanin-Spiral- und Ringstrukturen geben Aufschluss über den Kraftakt beim Abtrennen von Mikrotubuli. Nat. Struct. Mol. Biol. 24, 717-725. doi: 10.1038/nsmb.3448
PubMed Abstract | Querverweis Volltext | Google Scholar