der funktionelle Hauptbestandteil der roten Blutkörperchen, der als sauerstofftragendes Protein dient; es ist eine Art Hämoprotein, bei dem jedes Molekül ein Tetramer ist, das aus vier Monomeren besteht, die durch schwache Bindungen zusammengehalten werden. Es besteht aus zwei Paaren von Polypeptidketten, den Globinen, an die jeweils ein Häm-Molekül, bestehend aus Eisen und einem Protoporphyrin-Molekül, gebunden ist. Symbol Hb.
Das Eisenatom hat eine freie Valenz und kann ein Molekül Sauerstoff binden. Jedes Hämoglobinmolekül kann also ein Sauerstoffmolekül binden. Die Bindung von Sauerstoff durch ein Monomer erhöht die Sauerstoffaffinität der anderen im Tetramer. Dies macht Hämoglobin zu einem effizienteren Transportprotein als ein monomeres Protein wie Myoglobin.
Sauerstoffhaltiges Hämoglobin (Oxyhämoglobin) ist leuchtend rot; nicht an Sauerstoff gebundenes Hämoglobin (Desoxyhämoglobin) ist dunkler. Dies erklärt die hellrote Farbe des arteriellen Blutes, in dem das Hämoglobin zu etwa 97 Prozent mit Sauerstoff gesättigt ist. Venöses Blut ist dunkler, weil es nur zu etwa 20 bis 70 Prozent mit Sauerstoff gesättigt ist, je nachdem, wie viel Sauerstoff von den Geweben verbraucht wird. Die Affinität von Hämoglobin für Kohlenmonoxid ist 210-mal so stark wie seine Affinität für Sauerstoff. Der gebildete Komplex (Carboxyhämoglobin) kann keinen Sauerstoff transportieren. Eine Kohlenmonoxidvergiftung führt daher zu Hypoxie und Erstickung.
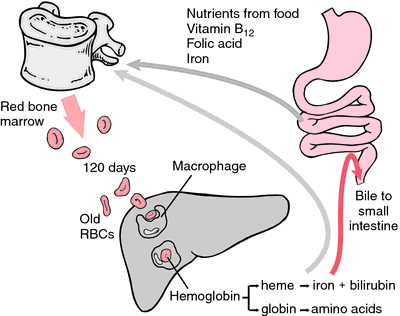
Eine andere Form von Hämoglobin, die keinen Sauerstoff transportieren kann, ist Methämoglobin, bei dem das Eisenatom zur Oxidationsstufe +3 oxidiert ist. Während der 120-tägigen Lebensdauer eines roten Blutkörperchens wird das Hämoglobin langsam zu Methämoglobin oxidiert. Mindestens vier verschiedene Enzymsysteme können Methämoglobin wieder in Hämoglobin umwandeln. Wenn diese Enzyme defekt oder überlastet sind, kann es zu einer Methämoglobinämie kommen, wobei hohe Methämoglobinwerte zu Atemnot und Zyanose führen.
Eine sekundäre Funktion von Hämoglobin ist die eines Teils des Blutpuffersystems. Die Histidinreste in den Globinketten wirken als schwache Basen, um die Veränderung des pH-Wertes im Blut zu minimieren, die bei der Aufnahme von Sauerstoff und der Freisetzung von Kohlendioxid in der Lunge sowie bei der Abgabe von Sauerstoff und der Aufnahme von Kohlendioxid aus dem Gewebe auftritt.
Wenn sich Erythrozyten abnutzen oder beschädigt werden, werden sie von den Makrophagen des retikuloendothelialen Systems aufgenommen. Der Porphyrinring des Häms wird in das Gallenpigment Bilirubin umgewandelt, das von der Leber ausgeschieden wird. Das Eisen wird zum Knochenmark transportiert, um in das Hämoglobin der neu gebildeten Erythrozyten eingebaut zu werden.
Die Hämoglobinkonzentration des Blutes variiert mit dem Hämatokrit. Die Normalwerte für die Hämoglobinkonzentration im Blut liegen bei 13,5 bis 18,0 g/100 ml bei Männern und 12,0 bis 16,0 g/100 ml bei Frauen. Die normale mittlere korpuskuläre Hämoglobinkonzentration, d. h. die Konzentration innerhalb der roten Blutkörperchen, beträgt 32 bis 36 g/100 ml.
Es wurden zahlreiche abnorme Hämoglobine entdeckt, die durch Mutationen entstanden sind. Einige haben eine veränderte Sauerstoffaffinität, andere sind instabil, und bei einigen ist das Eisenatom oxidiert, was zu einer kongenitalen Methämoglobinämie führt. Einige Mutationen führen zu einer verringerten Hämoglobinsyntheserate. Alle diese Erkrankungen werden als Hämoglobinopathien bezeichnet.
Die häufigste Hämoglobinopathie ist die Sichelzellkrankheit, die durch eine Mutation verursacht wird, bei der die sechste Aminosäure in der β-Kette, normalerweise Glutaminsäure, durch Valin ersetzt wird. Die Variante des Hämoglobins α2βS2 wird als Hb S bezeichnet. Mutationen, die zu einer verminderten Synthese einer der Ketten führen, werden Thalassämien genannt. Sie können durch eine Deletion des Gens für eine Kette oder durch eine Mutation in dem regulatorischen Gen, das die Synthese der Kette steuert, entstehen.