SONAL DESAI, ARCHITA PATEL UND S. Y. GABHE*
C. U. Shah College of Pharmacy, S. N. D. T. Women’s University, Sir Vithaldas Vidya Vihar, Juhu Tara Road, Santacruz (W), Mumbai – 400 049, India
Korrespondierender Autor: S. Y. GABHE E-mail:
| Datum der Annahme | 16-Jan-2011 |
| Datum der Überarbeitung | 26-Okt-2010 |
| Empfangsdatum | 9-Apr-2009 |
DOI: 10.4103/0250-474X.89762
Abstract
Eine einfache isokratische Umkehrphasen-Flüssigkeitschromatographie wurde verwendet, um drei Verunreinigungen in der Probe von 8-Chlorotheophyllin zu trennen. Für die Charakterisierung der Verunreinigungen wurde LC-MS verwendet. Anhand der Massenspektraldaten wurden die Strukturen dieser Verunreinigungen als 3,7-Dihydro-1,3-dimethyl-1H-purin-2,6-dion (Verunreinigung I), 3,7-Dihydro-1,3,7-trimethyl-1H-purin-2,6-dion (Verunreinigung II) und Isomer von 8-Chlor-1,3-dimethyl-2,6(3H,1H)-purindion (Verunreinigung III) charakterisiert.
Schlüsselwörter
8-Chlorotheophyllin, Verunreinigung, LC-MS, Umkehrphasen-HPLC, Theophyllin
Einführung
Die Bulk-Arzneimittelindustrie bildet die Grundlage aller pharmazeutischen Industrien, da sie die Quelle für pharmazeutische Wirkstoffe (APIs) von bestimmter Qualität ist. Die größte Herausforderung für die Bulk-Arzneimittelindustrie besteht darin, das fertige Arzneimittel in der erforderlichen Qualität und Reinheit wirtschaftlich herzustellen. Die Reinheit von Wirkstoffen hängt von mehreren Faktoren ab, wie z. B. von den Rohstoffen, ihren Herstellungsmethoden und der Art des Kristallisations- oder Reinigungsverfahrens. Es ist jedoch fast unmöglich, absolut reine Materialien zu erhalten, da Verunreinigungen entweder während der Herstellung, Reinigung oder Lagerung in die Materialien gelangen. Eine Verunreinigung ist jeder Bestandteil einer Arzneimittelsubstanz (mit Ausnahme von Wasser), der nicht zu der als Arzneimittelsubstanz definierten chemischen Einheit gehört. Bei den durch chemische Synthese hergestellten Arzneimitteln unterteilt die ICH die Verunreinigungen in drei Kategorien: organische Verunreinigungen, anorganische Verunreinigungen und Lösungsmittelrückstände. Die Verunreinigungen können toxisch sein, die physikalischen oder chemischen Eigenschaften der Substanz verändern und sie damit medizinisch unbrauchbar machen. Verunreinigungen können die Haltbarkeit des Produkts verkürzen und Schwierigkeiten bei der Formulierung verursachen. Daher ist nicht nur die Kontrolle von Verunreinigungen, sondern auch die Qualifizierung von Verunreinigungen für die Arzneimittelindustrie von entscheidender Bedeutung.
Antihistaminika wie Dimenhydrinat und Promethazintheoklat sind weit verbreitete Arzneimittel zur Behandlung der Reisekrankheit. 8-Chlorotheophyllin, chemisch gesehen 8-Chlor-1,3-dimethyl-2,6(1H, 3H)-purindion, ist ein Zwischenprodukt, das zur Herstellung der Salzform dieser Arzneimittel verwendet wird. Es ist wichtig, die Reinheit und Sicherheit von Dimenhydrinat und Promethazintheoklat zu gewährleisten. Um dies zu erreichen, muss 8-Chlortheophyllin in höchster Reinheit und mit einem bekannten Verunreinigungsprofil gewonnen werden.
Aus der Literaturübersicht geht hervor, dass Wadke et al. die Wechselwirkungen von 9-Methylisoalloxazin und 3,9-Dimethylisoalloxazin mit 8-Chlortheophyllin untersuchten. 8-Chlortheophyllin wurde als interner Standard für die Bestimmung von Urat mittels HPLC-Methode verwendet. Es wurde auch potentiometrisch bestimmt. Die gleichzeitige Bestimmung von Chlorphenoxaminhydrochlorid, 8-Chlortheophyllin und Koffein in einer Mehrkomponenten-Darreichungsform wurde mit einer dünnschichtchromatographisch-densitometrischen Methode durchgeführt. Gil et al. untersuchten das elektroanalytische Verhalten von 8-Chlortheophyllin mittels zyklischer Voltammetrie und differentieller Pulspolarographie und bestimmten seinen Gehalt in pharmazeutischen Zubereitungen mittels differentieller Pulspolarographie. Eine stabilitätsanzeigende RP-HPLC-Methode wurde für 8-Chlortheophyllin zusammen mit Diphenhydramin und Koffein entwickelt und validiert. Für die gleichzeitige Bestimmung von Koffein, 8-Chlortheophyllin und Chlorphenoxamin-Hydrochlorid in ternären Mischungen wurden außerdem spektrophotometrische und chemometrische Methoden mit Nulldurchgang entwickelt. Bislang wurde keine chromatographische und spektroskopische Methode zur Trennung und Charakterisierung von Verunreinigungen in 8-Chlortheophyllin beschrieben. Daher wurde die vorliegende Arbeit mit dem Ziel durchgeführt, Verunreinigungen in 8-Chlorotheophyllin mit Hilfe moderner analytischer Techniken zu isolieren und zu charakterisieren.
8-Chlorotheophyllin war eine Geschenkprobe von Kores (India) Ltd. in Thane. Alle anderen Chemikalien und Reagenzien wurden von S. D. Fine Chemicals Ltd (Mumbai, Indien) beschafft. Die für TLC- und präparative TLC-Studien verwendeten Lösungsmittel waren von analytischer Qualität und die für HPLC-Studien verwendeten von HPLC-Qualität. Natriumacetat-Trihydrat von AR-Qualität wurde für die Vorbereitung der Puffer verwendet.
Zunächst wurden TLC-Studien durchgeführt, um die Anzahl der in der Probe vorhandenen Verunreinigungen zu ermitteln. Als stationäre Phase wurden vorbeschichtete TLC-Platten aus Kieselgel 60GF254 (Merck) verwendet. Die Probe wurde in einer minimalen Menge Ethylacetat gelöst, und diese Lösung wurde zum Tupfen der TLC-Platten verwendet. Es wurden verschiedene mobile Phasen ausprobiert. Ethylacetat:Toluol:Eisessig (10:0,3:0,5 v/v/v) zeigte im Vergleich zu anderen mobilen Phasen eine bessere Trennung. Aus der Probe von 8-Chlorotheophyllin wurden mittels TLC vier Komponenten mit den Rf-Werten von 0,029, 0,132, 0,198 bzw. 0,852 abgetrennt. 8-Chlorotheophyllin hatte einen Rf-Wert von 0,852.
Nachdem die mobile Phase für die TLC entwickelt worden war, wurde versucht, das Gemisch mit präparativer TLC zu trennen. Die Probe wurde in einer minimalen Menge Ethylacetat gelöst und in Form einer Bande aufgetragen. Alle drei Verunreinigungen wurden durch präparative TLC von 8-Chlorotheophyllin getrennt, wobei die gleichen chromatographischen Bedingungen wie bei den TLC-Studien verwendet wurden. Die verschiedenen Banden wurden getrennt gesammelt und mit Ethylacetat extrahiert. Da die Mengen der einzelnen isolierten Verunreinigungen I, II und III sehr gering waren, wurde beschlossen, diese Verunreinigungen gemeinsam zu isolieren. Die Verunreinigungen I, II und III wurden zusammen als Fraktion I bezeichnet. Die Fraktion I wurde durch präparative TLC aus 8-Chlorotheophyllin isoliert. Da die Verunreinigungen nicht einzeln isoliert wurden, konnten verschiedene Identifizierungstechniken wie IR und NMR nicht durchgeführt werden. Es wurde beschlossen, weitere Untersuchungen mittels LC-MS durchzuführen, die eine gleichzeitige Trennung und Charakterisierung ermöglicht. Vor der LC-MS-Analyse wurde ein HPLC-Profil für die Fraktion I erstellt. Für die HPLC-Untersuchungen wurde ein Hochleistungsflüssigkeitschromatograph von Tosoh verwendet, der mit einer CCPM-Kolbenpumpe, einer Pumpensteuerung PX8010 und einem UV-Detektor ausgestattet war. Eine Schleife mit einem Fassungsvermögen von 20 µl wurde an die Injektionsventileinheit angeschlossen. Fraktion I wurde in Acetonitril aufgelöst und einer Umkehrphasen-HPLC-Analyse mit einer mobilen Phase aus Acetonitril: Natriumacetat-Trihydrat (pH 3,57; 0,01 M) (5:95 v/v) unterzogen. Die gewählte Säule war eine Phenomenex ODS (250×4,6 mm I.D.; Partikelgröße 5 µm). Die Durchflussrate betrug 1,5 ml/min und die Detektion wurde bei einer Wellenlänge von 280 nm überwacht. Die mobile Phase wurde vor der Verwendung durch einen G5-Sinterglasfilter unter Vakuum filtriert und mit Ultraschall von Luftblasen befreit. Die HPLC-Analyse der Fraktion I ergab ebenfalls drei Peaks mit einer mittleren Retentionszeit von 8,324 min, 14,275 min bzw. 20,933 min (Abb. 1). Der Peak mit einer mittleren Retentionszeit von 2,086 min war der Lösungsmittelpeak.
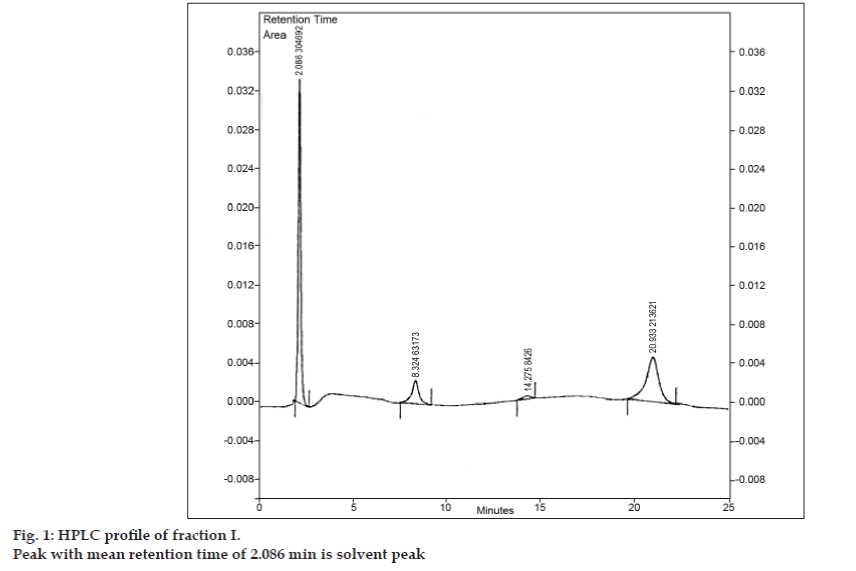
Abbildung 1: HPLC-Profil der Fraktion I.
Die Fraktion I wurde anschließend einer LC-MS-Analyse zur Charakterisierung der Verunreinigungen unterzogen. Die LC-MS-Untersuchungen wurden mit einem System durchgeführt, bei dem der LC-Teil aus einer HPLC der Serie 1100 (Agilent Technologies, USA) mit einem Vakuumentgaser (G1322A), einer quaternären Pumpe (G1311A), einem Auto-Sampler (G1313A) und einem UV/sichtbaren Detektor (G1314A) und der MS-Teil aus einem Triple-Quadrupol-Massenspektrometer Quattro II (Micromass UK Ltd.,
In den LC-MS-Studien wurden die flüssigchromatographischen Trennungen mit einer Phenomenex C18-Säule (250×4,6 mm, 5 µm) bei Raumtemperatur und einer mobilen Phase aus Acetonitril: Natriumacetat-Puffer (5:95, v/v) mit einer Flussrate von 1,5 ml/min durchgeführt. Das Massenspektrometer wurde im ESI-Modus (positive electron spray ionization) mit einem Masse/Ladungs-Verhältnis (m/z) im Bereich von 120-500 m/z betrieben. Als Vernebelungsgas wurde Stickstoff verwendet. Die Daten wurden mit der Software Masslynx erfasst und verarbeitet. Es wurden Massenspektraldaten der Verunreinigungen erhalten (Abb. 2, 3 und 4). Der Fragmentierungsweg für drei Peaks ist durch den Verlust der Methylgruppe und/oder der Carbonylgruppe gekennzeichnet. Die Peaks mit m/z-Werten von 180,9, 195,7 und 214,9 entsprechen den +, + bzw. +-Peaks. Den MS-Daten zufolge handelte es sich bei den Verunreinigungen, die bei 8,324 min, 14,275 min und 20,933 min eluierten, um Theophyllin (Molgewicht 180), Koffein (Molgewicht 194) bzw. ein Isomer von 8-Chlorotheophyllin (Molgewicht 214,5) (Tabelle 1). So wurden drei Verunreinigungen abgetrennt und ihre Strukturen anhand von Massenspektraldaten aufgeklärt (Schema 1).
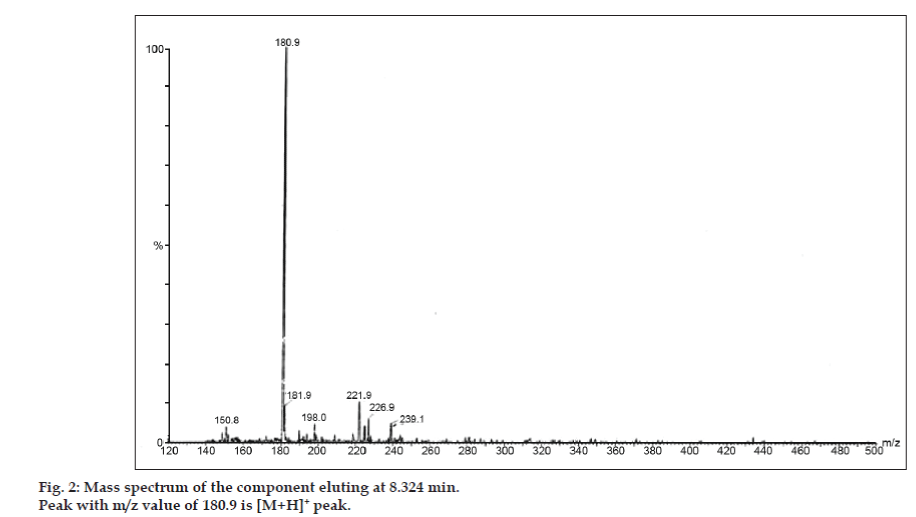
Abb. 2: Massenspektrum der bei 8,324 min eluierenden Komponente.
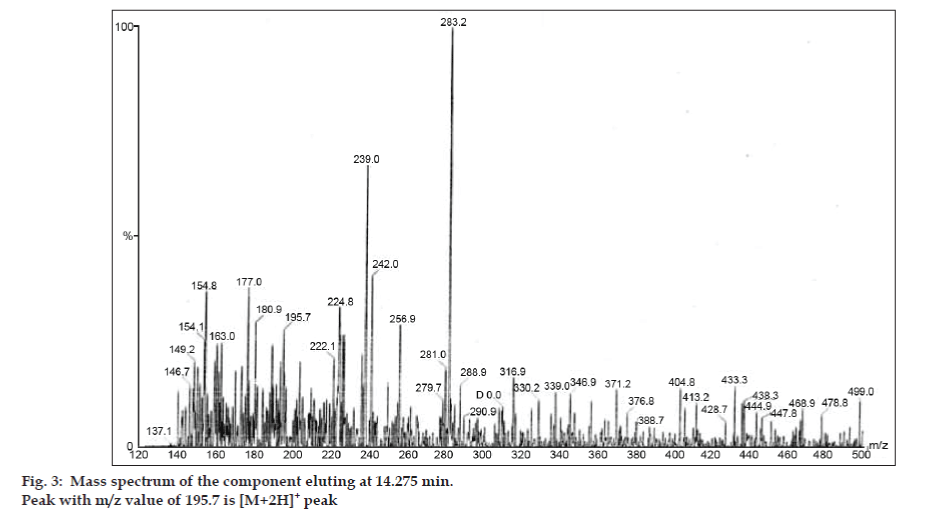
Abb. 3: Massenspektrum der Komponente, die bei 14,275 min eluiert.
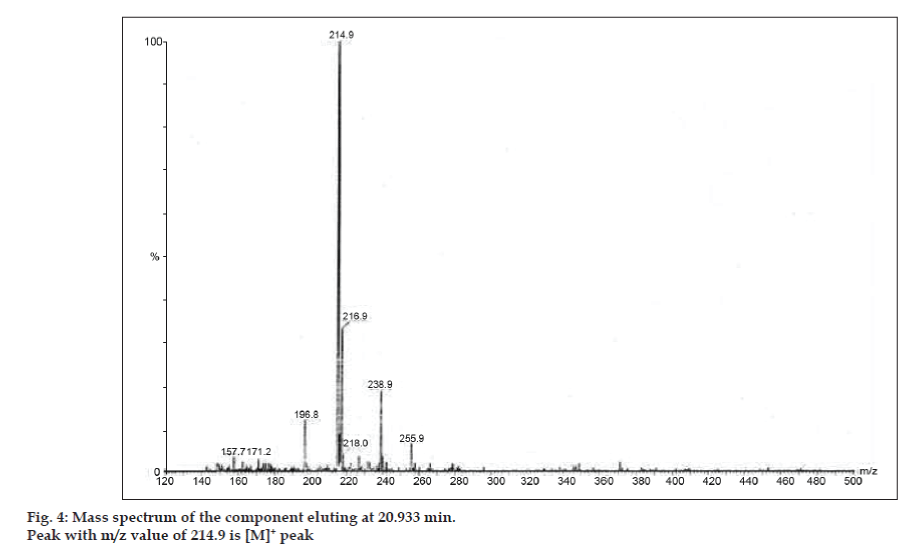
Abbildung. 4: Massenspektrum der bei 20,933 min eluierenden Komponente.
| Peak Nr. |
Retentionszeit (min) |
Fragment-Ionen (m/z) | Identifikation |
|---|---|---|---|
| 8.324 | 181.9 +, 180.9 +, 150.8 + | Theophyllin | |
| 14.275 | 195.7 +, 180.9 +,149.2 +, 137.1+ | Koffein | |
| 20.933 | 216.9 +, 214.9 +, 171.2 +, 157.7 + | Isomer von 8- Chlorotheophyllin |
TABELLE 1: HPLC-MS IDENTIFIZIERUNG DER FRACTION I
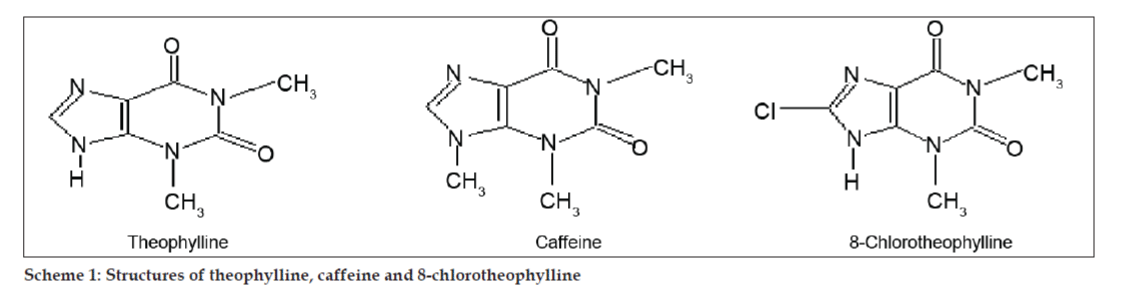
Schema 1: Strukturen von Theophyllin, Koffein und 8-Chlorotheophyllin
Danksagungen
Die Autoren danken Kores (India) Ltd, Thane für die Bereitstellung einer Probe von 8-Chlortheophyllin.
- Kasture AV, Wadodkar SG, Mahadik KR, More HV. Pharmaceutical Analysis. Vol. 1. Pune: NiraliPrakashan; 1997. p. 12-4.
- United State Pharmacopoeia, Vol. 26. Rockville, MD: United States Pharmacopoeia Convention, Inc.; 1999. p. 2049-59.
- Ahuja S, Alsante KM. Handbook of isolation and characterization of impurities in pharmaceuticals. California: Academic Press; 2003. p. 6.
- Foye WO, Lemke TL, Williams DA. Principles of Medicinal Chemistry. 4th ed. New Delhi: B. I. Waverly Pvt Ltd; 1995. p. 419.
- Reynolds JE. Martindale-The Extra Pharmacopoeia. 29th ed. London: Pharmaceutical Press; 1989. p. 451,459.
- Wadke DA, Guttman DE. Complex formation influence on reaction rate III. Interaction of some isoalloxazines with 8-chlorotheophylline as determined by spectral, solubility, and kinetic methods. J Pharm Sci 1965;54:1293.
- Bennett MJ, Patchett BP, Worthy E. A simple HPLC method for the determination of urate in serum and urine using 8-chlorotheophylline as internal standard. Med Lab Sci 1984;41:108-11.
- Nikolic K, Medenica M. Potentiometric determination of 8-chlorotheophylline. Microchimica Acta 1986;88:5.
- Bebawy LI, El-Kousy NM. Simultane Bestimmung einiger Multikomponenten-Darreichungsformen mittels quantitativer Dünnschichtchromatographie-Densitometriemethode. J Pharm Biomed Anal 1999;20:663-70.
- Gil EP, Blazquez LC, Garcia-MoncoCarra RM, Misiego AS. Polarographic Behavior of 8-Chlorotheophylline and its Determination in Dosierungsformen. Electroanalysis 1993;5:343.
- Barbas C, Garcia A, Saavedra L, Castro M. Optimization and validation of a method for the determination of caffeine, 8-chlorotheophylline and diphenhydramine by isocratic high-performance liquid chromatography Stress test for stability evaluation. J ChromatogrA 2000;870:97-103.
- Kelani KM. Gleichzeitige Bestimmung von Koffein, 8-Chlortheophyllin und Chlorphenoxaminhydrochlorid in ternären Mischungen durch verhältnisspektrometrische, nulldurchgangsbestimmende, erstableitende spektrophotometrische und chemometrische Methoden. J AOAC Int 2005;88:1126-34.