SONAL DESAI, ARCHITA PATEL Y S. Y. GABHE*
C. U. Shah College of Pharmacy, S. N. D. T. Women’s University, Sir Vithaldas Vidya Vihar, Juhu Tara Road, Santacruz (W), Mumbai – 400 049, India
Autor correspondiente: S. Y. GABHE E-mail:
| Fecha de aceptación | 16-Enero-2011 |
| Fecha de revisión | 26-Oct-2010 |
| Fecha de recepción | 9-Abr-2009 |
DOI: 10.4103/0250-474X.89762
Abstracto
Se utilizó una cromatografía líquida de alto rendimiento isocrática simple para separar tres impurezas presentes en la muestra de 8-cloroteofilina. Se utilizó LC-MS para la caracterización de las impurezas. Basándose en los datos del espectro de masas, las estructuras de estas impurezas se caracterizaron como 3,7-dihidro-1,3-dimetil-1H-purina-2,6-diona (impureza I), 3,7-dihidro-1,3,7-trimetil-1H-purina-2,6-diona (impureza II) e isómero de 8-cloro-1,3-dimetil-2,6(3H,1H)-purinediona (impureza III).
Palabras clave
8-clorofilina, impureza, LC-MS, HPLC de fase inversa, teofilina
Introducción
La industria de medicamentos a granel constituye la base de todas las industrias farmacéuticas, ya que es la fuente de ingredientes farmacéuticos activos (API) de calidad especificada. El mayor reto para las industrias de medicamentos a granel es producir el medicamento final con la calidad y pureza requeridas, de forma económica. La pureza de los API depende de varios factores, como las materias primas, sus métodos de fabricación y el tipo de proceso de cristalización o purificación. Sin embargo, es casi imposible obtener materiales absolutamente puros, ya que las impurezas se incorporan a ellos durante la fabricación, la purificación o el almacenamiento. Una impureza es cualquier componente de una sustancia farmacológica (excluyendo el agua) que no es una entidad química definida como sustancia farmacológica. En el caso de los medicamentos producidos por síntesis química, la ICH clasifica las impurezas en tres categorías: impurezas orgánicas, impurezas inorgánicas y disolventes residuales. Las impurezas presentes en la sustancia pueden ser tóxicas, pueden cambiar las propiedades físicas o químicas de la sustancia, haciéndola inútil desde el punto de vista medicinal. Las impurezas pueden reducir la vida útil del producto y causar dificultades en la formulación. Por lo tanto, no sólo el control de las impurezas, sino también su calificación, es una cuestión crítica para las industrias de medicamentos a granel.
Los antihistamínicos, como el dimenhidrinato y el teocato de prometazina, son fármacos muy utilizados en el tratamiento del mareo. La 8-clorofilina, químicamente 8-cloro-1,3-dimetil-2,6(1H, 3H)- purinediona, es un intermedio que se utiliza en la preparación de la forma salina de estos fármacos. Es esencial garantizar la pureza y la seguridad del dimenhidrinato y del teoclato de prometazina. Para ello, la 8-cloroteofilina debe obtenerse con la máxima pureza y con un perfil de impurezas conocido.
De la investigación bibliográfica se desprende que Wadke et al. estudiaron las interacciones de la 9-metilisoaloxazina y la 3,9-dimetilisoaloxazina con la 8-cloroteofilina. La 8-cloroteofilina se utilizó como estándar interno para la determinación del urato por el método HPLC. También se determinó potenciométricamente. La determinación simultánea del clorhidrato de clorfenoxamina, la 8-cloroteofilina y la cafeína en la forma farmacéutica multicomponente se llevó a cabo mediante un método de cromatografía en capa fina-densitométrica. Gil et al. investigaron el comportamiento electroanalítico de la 8-cloroteofilina por voltametría cíclica y por polarografía diferencial de impulsos y determinaron su contenido en preparados farmacéuticos por polarografía diferencial de impulsos. Se desarrolló y validó un método RP-HPLC indicador de estabilidad para la 8-cloroteofilina junto con la difenhidramina y la cafeína. También se desarrollaron métodos espectrofotométricos y quimiométricos de relación-espectro y cruce de cero para la determinación simultánea de cafeína, 8-cloroteofilina y clorofenoxamina en mezclas ternarias. Hasta ahora no se ha informado de ningún método cromatográfico y espectroscópico para la separación y caracterización de las impurezas presentes en la 8-cloroteofilina. Por lo tanto, el presente trabajo se llevó a cabo con el objetivo de aislar y caracterizar las impurezas presentes en la 8-cloroteofilina utilizando técnicas analíticas modernas.
La 8-cloroteofilina fue una muestra regalada por Kores (India) Ltd., Thane. Todos los demás productos químicos y reactivos se obtuvieron de S. D. Fine Chemicals Ltd (Mumbai, India). Los disolventes utilizados para los estudios de TLC y TLC preparativa eran de grado analítico y los utilizados para los estudios de HPLC eran de grado HPLC. Se utilizó acetato de sodio trihidratado de grado AR para la preparación del tampón.
Inicialmente se realizaron estudios de TLC para conocer el número de impurezas presentes en la muestra. Como fase estacionaria se utilizaron placas de TLC precubiertas de gel de sílice 60GF254 (Merck). La muestra se disolvió en una cantidad mínima de acetato de etilo y esta solución se utilizó para manchar las placas de TLC. Se probaron varias fases móviles. Acetato de etilo:tolueno:ácido acético glacial (10:0,3:0,5 v/v/v) mostró una mejor separación en comparación con otras fases móviles. Se separaron cuatro componentes de la muestra de 8-cloroteofilina mediante TLC con los valores Rf de 0,029, 0,132, 0,198 y 0,852, respectivamente. La 8-cloroteofilina tenía un Rf de 0,852.
Una vez desarrollada la fase móvil para la TLC, se intentó separar la mezcla utilizando la TLC preparatoria. La muestra se disolvió en una cantidad mínima de acetato de etilo y se manchó en forma de banda. Las tres impurezas se separaron de la 8-clorofilina mediante TLC preparativa utilizando las mismas condiciones cromatográficas empleadas en los estudios de TLC. Las diferentes bandas se recogieron por separado y se extrajeron utilizando acetato de etilo. Dado que las cantidades de cada impureza aislada I, II y III eran muy reducidas, se decidió aislar estas impurezas colectivamente. Las impurezas I, II y III se designaron colectivamente como fracción I. La fracción I se aisló de la 8-clorofilina mediante TLC preparativa. Como cada impureza no se aisló por separado, no se pudieron llevar a cabo diferentes técnicas de identificación como IR, NMR. Se decidió llevar a cabo una investigación adicional mediante LC-MS, que permite la separación y caracterización simultáneas. Antes del análisis LC-MS, se desarrolló un perfil HPLC para la fracción I. Para los estudios de HPLC se utilizó un cromatógrafo líquido de alto rendimiento Tosoh equipado con una bomba reciprocante CCPM, un controlador de bomba PX8010 y un detector UV. Se colocó un bucle de 20 µl de capacidad en la unidad de válvula de inyección. La fracción I se disolvió en acetonitrilo y se sometió a un análisis de HPLC de fase inversa con fase móvil compuesta por acetonitrilo: acetato de sodio trihidratado (pH 3,57; 0,01 M) (5:95 v/v). La columna seleccionada fue la Phenomenex ODS (250×4,6 mm I.D.; tamaño de partícula 5 µm). El caudal fue de 1,5 ml/min y la detección se controló a una longitud de onda de 280 nm. La fase móvil se filtró a través de un filtro de vidrio sinterizado G5 bajo vacío antes de su uso y se sonicó para eliminar las burbujas de aire. El análisis por HPLC de la fracción I también reveló tres picos con tiempos de retención medios de 8,324 min, 14,275 min y 20,933 min, respectivamente (fig. 1). El pico con un tiempo de retención medio de 2,086 min era el pico del disolvente.
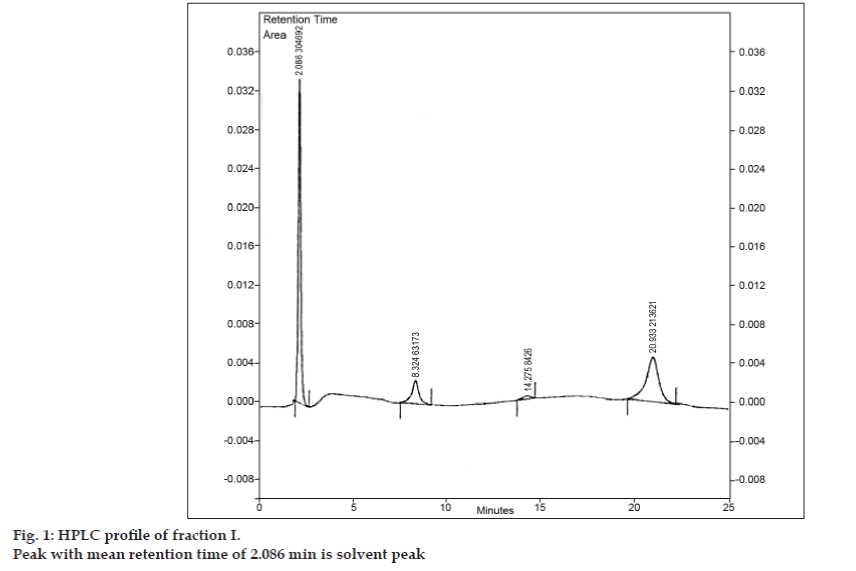
Figura 1: Perfil de HPLC de la fracción I.
La fracción I se sometió entonces a un análisis de LC-MS para la caracterización de las impurezas. Los estudios de LC-MS se llevaron a cabo en un sistema en el que la parte de LC consistía en un HPLC de la serie 1100 (Agilent Technologies, EE.UU.) que comprendía un desgasificador al vacío (G1322A), una bomba cuaternaria (G1311A), un muestreador automático (G1313A) y un detector UV/visible (G1314A) y la parte de MS consistía en un espectrómetro de masas de triple cuadrupolo Quattro II (Micromass UK Ltd., Reino Unido).
En los estudios de LC-MS, las separaciones cromatográficas líquidas se lograron con la columna Phenomenex C18 (250×4,6 mm, 5 µm) a temperatura ambiente con la fase móvil de acetonitrilo: tampón de acetato de sodio (5:95, v/v) a un caudal de 1,5 ml/min. El espectrómetro de masas funcionó en modo de ionización por pulverización de electrones (ESI) positiva con una relación masa/carga (m/z) en el rango de 120-500 m/z. Se utilizó nitrógeno como gas nebulizador. Los datos se adquirieron y procesaron con el software Masslynx. Se obtuvieron datos espectrales de masas de las impurezas (figs. 2, 3 y 4). La vía de fragmentación de tres picos se caracteriza por la pérdida del grupo metilo y/o del grupo carbonilo. Los picos con valores m/z de 180,9, 195,7 y 214,9 corresponden a picos +, + y + respectivamente. Según los datos de EM obtenidos, las impurezas que eluyen a los 8,324 min, 14,275 min y 20,933 min son la teofilina (peso mol. 180), la cafeína (peso mol. 194) y un isómero de la 8-clorofilina (peso mol. 214,5), respectivamente (Tabla 1). Así, se separaron tres impurezas y se dilucidaron sus estructuras a partir de los datos del espectro de masas (esquema 1).
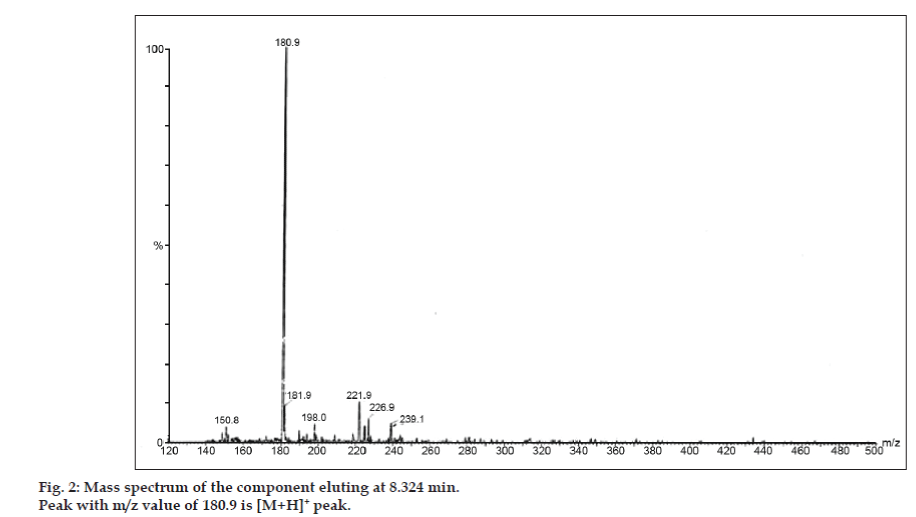
Fig 2: Espectro de masas del componente que eluye a 8,324 min.
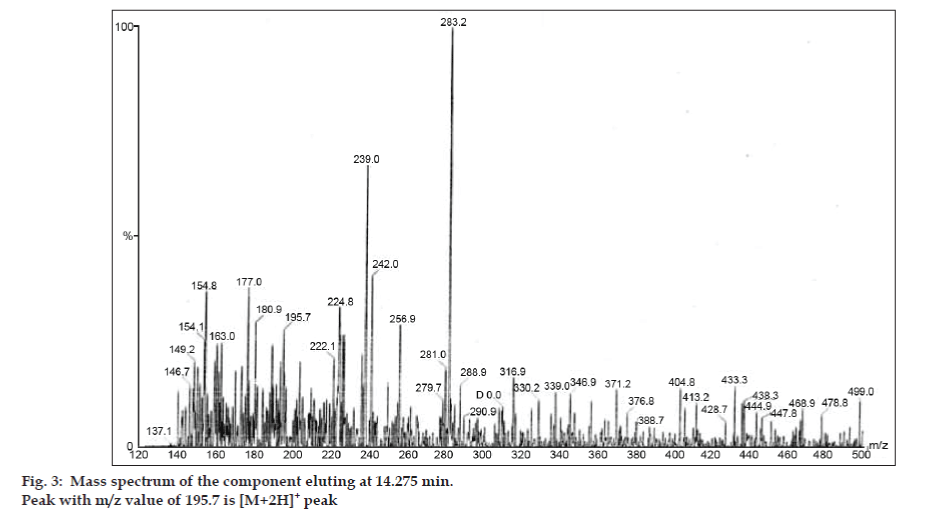
Fig. 3: Espectro de masas del componente que eluye a 14,275 min.
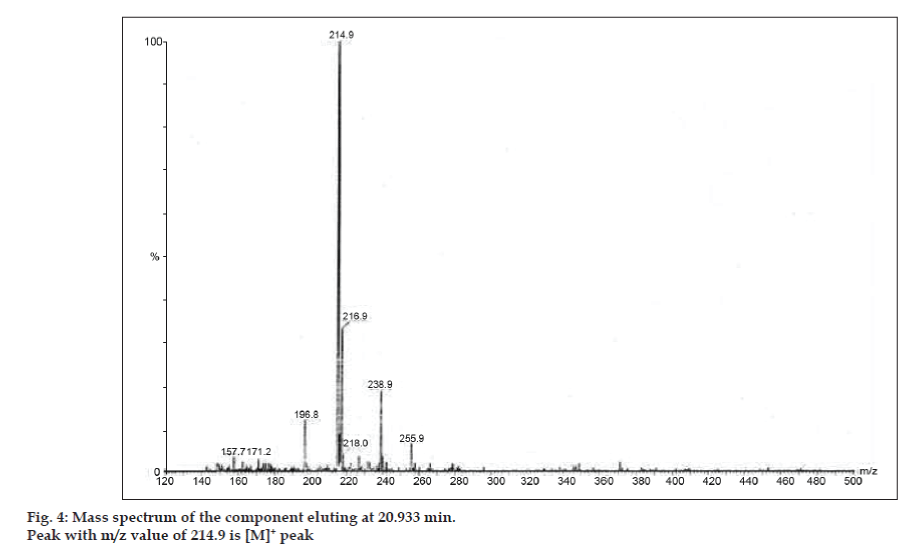
Fig. 4: Espectro de masas del componente que eluye a 20,933 min.
| Pico no. |
Tiempo de retención (min) |
Iones de fragmento (m/z) | Identificación |
|---|---|---|---|
| 8.324 | 181,9 +, 180,9 +, 150,8 + | Teofilina | |
| 195,7 +, 180,9 +,149,2 +, 137,1+ | Café | ||
| 20,933 | 216.9 +, 214.9 +, 171.2 +, 157.7 + | Isómero de la 8- Clorofilina |
TABLA 1: IDENTIFICACIÓN POR HPLC-MS DE LA FRACCIÓN I
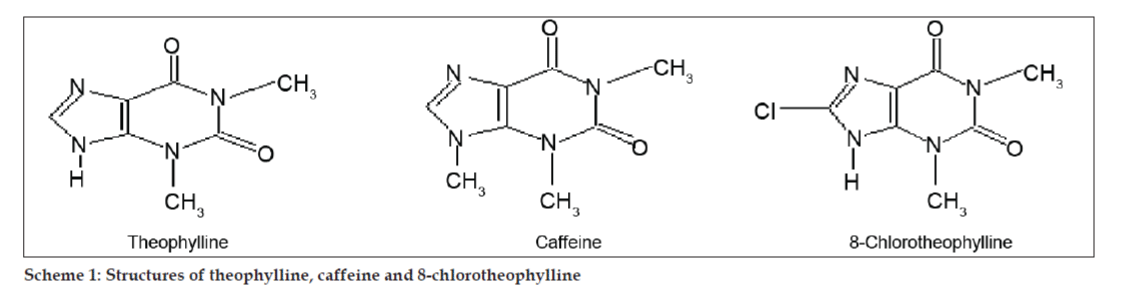
Esquema 1: Estructuras de la teofilina, la cafeína y la 8-cloroteofilina
Agradecimientos
Los autores agradecen a Kores (India) Ltd., Thane por proporcionar una muestra de regalo de 8-clorofilina.
- Kasture AV, Wadodkar SG, Mahadik KR, More HV. Pharmaceutical Analysis. Vol. 1. Pune: NiraliPrakashan; 1997. p. 12-4.
- United State Pharmacopoeia, Vol. 26. Rockville, MD: United States Pharmacopoeia Convention, Inc.; 1999. p. 2049-59.
- Ahuja S, Alsante KM. Handbook of isolation and characterization of impurities in pharmaceuticals. California: Academic Press; 2003. p. 6.
- Foye WO, Lemke TL, Williams DA. Principles of Medicinal Chemistry. 4th ed. New Delhi: B. I. Waverly Pvt Ltd; 1995. p. 419.
- Reynolds JE. Martindale-The Extra Pharmacopoeia. 29th ed. London: Pharmaceutical Press; 1989. p. 451,459.
- Wadke DA, Guttman DE. Influencia de la formación de complejos en la velocidad de reacción III. Interacción de algunas isoaloxazinas con la 8-clorofilina determinada por métodos espectrales, de solubilidad y cinéticos. J Pharm Sci 1965;54:1293.
- Bennett MJ, Patchett BP, Worthy E. A simple HPLC method for the determination of urate in serum and urine using 8-chlorotheophylline as internal standard. Med Lab Sci 1984;41:108-11.
- Nikolic K, Medenica M. Potentiometric determination of 8-chlorotheophylline. Microchimica Acta 1986;88:5.
- Bebawy LI, El-Kousy NM. Determinación simultánea de algunas formas de dosificación multicomponente por método densitométrico de cromatografía en capa fina cuantitativa. J Pharm Biomed Anal 1999;20:663-70.
- Gil EP, Blazquez LC, Garcia-MoncoCarra RM, Misiego AS. Comportamiento polarográfico de la 8-clorofilina y su determinación en formas farmacéuticas. Electroanálisis 1993;5:343.
- Barbas C, García A, Saavedra L, Castro M. Optimización y validación de un método para la determinación de cafeína, 8-cloroteofilina y difenhidramina por cromatografía líquida isocrática de alta resolución Prueba de estrés para la evaluación de la estabilidad. J ChromatogrA 2000;870:97-103.
- Kelani KM. Determinación simultánea de cafeína, 8-clorofilina y clorofenoxamina hidrocloruro en mezclas ternarias por métodos espectrofotométricos y quimiométricos de relación-espectro de cruce de cero. J AOAC Int 2005;88:1126-34.