el principal constituyente funcional de los glóbulos rojos, que sirve como proteína transportadora de oxígeno; es un tipo de hemoproteína en la que cada molécula es un tetrámero compuesto por cuatro monómeros unidos por enlaces débiles. Está formada por dos pares de cadenas polipeptídicas, las globinas, cada una de las cuales tiene una molécula de hemo unida, compuesta de hierro más una molécula de protoporfirina. Símbolo Hb.
El átomo de hierro tiene una valencia libre y puede unir una molécula de oxígeno. Así, cada molécula de hemoglobina puede unir una molécula de oxígeno. La unión del oxígeno por parte de un monómero aumenta la afinidad por el oxígeno de los demás en el tetrámero. Esto hace que la hemoglobina sea una proteína de transporte más eficiente que una proteína monomérica como la mioglobina.
La hemoglobina oxigenada (oxihemoglobina) es de color rojo brillante; la hemoglobina no unida al oxígeno (desoxihemoglobina) es más oscura. Esto explica el color rojo brillante de la sangre arterial, en la que la hemoglobina está saturada de oxígeno en un 97%. La sangre venosa es más oscura porque sólo está saturada entre un 20% y un 70%, dependiendo de la cantidad de oxígeno que utilicen los tejidos. La afinidad de la hemoglobina por el monóxido de carbono es 210 veces mayor que su afinidad por el oxígeno. El complejo formado (carboxihemoglobina) no puede transportar oxígeno. Así, la intoxicación por monóxido de carbono provoca hipoxia y asfixia.
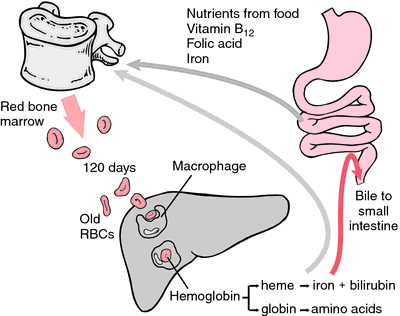
Otra forma de hemoglobina que no puede transportar oxígeno es la metahemoglobina, en la que el átomo de hierro está oxidado al estado de oxidación +3. Durante los 120 días de vida de un glóbulo rojo, la hemoglobina se oxida lentamente a metahemoglobina. Al menos cuatro sistemas enzimáticos diferentes pueden convertir la metahemoglobina en hemoglobina. Cuando éstos son defectuosos o están sobrecargados, puede producirse una metahemoglobinemia, en la que los niveles elevados de metahemoglobina provocan disnea y cianosis.
Una función secundaria de la hemoglobina es formar parte del sistema de amortiguación de la sangre. Los residuos de histidina en las cadenas de globina actúan como bases débiles para minimizar el cambio en el pH de la sangre que se produce a medida que se absorbe el oxígeno y se libera el dióxido de carbono en los pulmones y a medida que se suministra el oxígeno y se absorbe el dióxido de carbono de los tejidos.
A medida que los eritrocitos se desgastan o se dañan, son ingeridos por los macrófagos del sistema reticuloendotelial. El anillo de porfirina del hemo se convierte en el pigmento biliar bilirrubina, que es excretado por el hígado. El hierro se transporta a la médula ósea para incorporarse a la hemoglobina de los eritrocitos recién formados.
La concentración de hemoglobina de la sangre varía con el hematocrito. Los valores normales de la concentración de hemoglobina en sangre son de 13,5 a 18,0 g/100 ml en los hombres y de 12,0 a 16,0 g/100 ml en las mujeres. La concentración media normal de hemoglobina corpuscular, que es la concentración dentro de los glóbulos rojos, es de 32 a 36 g/100 ml.
Se han descubierto muchas hemoglobinas anormales derivadas de mutaciones. Algunas tienen una afinidad al oxígeno alterada, otras son inestables y en algunas el átomo de hierro está oxidado, lo que da lugar a una metahemoglobinemia congénita. Algunas mutaciones dan lugar a una tasa reducida de síntesis de hemoglobina. Todas estas condiciones se conocen como hemoglobinopatías.
La hemoglobinopatía más común es la enfermedad de células falciformes, causada por una mutación que sustituye el sexto aminoácido de la cadena β, normalmente ácido glutámico, por valina. La variante de la hemoglobina α2βS2 se conoce como Hb S. Las mutaciones que dan lugar a una síntesis reducida de una de las cadenas se denominan talasemias. Pueden resultar de la deleción del gen de una cadena o de una mutación en el gen regulador que controla la síntesis de la cadena.