Editorial sur le sujet de recherche
Le rôle des protéines AAA+ dans la réparation et la dégradation des protéines. Réparation et dégradation des protéines
Les ATPases associées à diverses activités cellulaires (AAA+) comprennent une superfamille de protéines qui remplissent une grande variété de fonctions essentielles à la physiologie cellulaire, notamment le contrôle de l’homéostasie des protéines, la réplication de l’ADN, la recombinaison, le remodelage de la chromatine, le traitement de l’ARN ribosomal, le ciblage moléculaire, la biogenèse des organelles et la fusion membranaire (Hanson et Whiteheart, 2005 ; Erzberger et Berger, 2006 ; Snider et al., 2008). Les membres de cette superfamille sont définis par la présence de ce que l’on appelle le domaine AAA+, qui contient les motifs canoniques Walker A et B nécessaires à la liaison et à l’hydrolyse de l’ATP (Hanson et Whiteheart, 2005). En général, les génomes codent pour environ dix à plusieurs centaines de membres de la famille AAA+ (tableau 1 ; Finn et al., 2017), chacun d’entre eux étant censé être adapté à des niches fonctionnelles spécifiques qui nécessitent des mécanismes précis de reconnaissance et de traitement du substrat (Hanson et Whiteheart, 2005). Le rayonnement adaptatif frappant des protéines AAA+ pour opérer dans divers contextes illustre l’utilité polyvalente du domaine AAA+ (Erzberger et Berger, 2006). Les protéines AAA+ forment généralement des complexes hexamériques et agissent comme des moteurs pour remodeler d’autres protéines, de l’ADN/ARN ou des complexes multicomposants (Figure 1). En effet, de nombreuses chaperonnes et protéases ATP-dépendantes sont ou ont des sous-unités qui appartiennent à cette superfamille (Figure 1 ; Olivares et al., 2016).
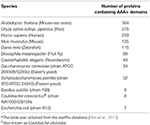
Tableau 1. Nombre de protéines AAA+ dans les organismes modèlesa.
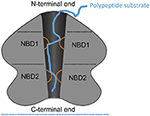
Figure 1. Schéma de ClpA comme exemple d’hexamère AAA+. Représentation schématique de l’architecture de domaine et des interactions de ClpA comme un exemple d’hexamère AAA+ avec deux domaines AAA+ par monomère. Ici, une vue latérale coupée de l’hexamère ClpA est montrée pour illustrer le canal central conducteur de polypeptide. ClpA contient trois domaines, dont un domaine N-terminal et deux domaines AAA+ : les domaines de liaison aux nucléotides 1 et 2 (NBD1 et NBD2). Le domaine N-terminal interagit avec les régulateurs de la spécificité du substrat, tandis que l’extrémité C-terminale interagit avec la protéase à chambre ClpP. Les boucles mobiles de NBD1 et NBD2 (orange) se projettent dans le canal central et engagent le substrat polypeptidique (bleu) permettant ainsi le couplage de l’hydrolyse de l’ATP à la translocation du polypeptide à travers le canal central.
Au cours des dernières années, des progrès substantiels ont été réalisés dans l’identification de la structure et du mécanisme fonctionnel d’un grand nombre de protéines AAA+ (Gates et al., 2017 ; Puchades et al., 2017 ; Ripstein et al., 2017 ; Zehr et al., 2017). Dans ce sujet de recherche, plusieurs éléments de ces progrès passionnants sont transmis dans 21 articles, qui englobent une vue structurelle et mécanistique détaillée de plusieurs chaperons et protéases AAA+, notamment : ClpX (Alhuwaider et Dougan ; Bittner et al. ; Elsholz et al. ; LaBreck et al. ; Vass et al.), ClpA (Bittner et al. ; Duran et al.), ClpB et Hsp104 (Chang et al.; Duran et al. ; Franke et al. ; Johnston et al.), Hsp78 (Abrahão et al.), ClpC (Alhuwaider et Dougan ; Elsholz et al.), ClpE (Elsholz et al.), Pontin (Mao et Houry), Reptin (Mao et Houry), FtsH (Alhuwaider et Dougan), protéasome 19S (Snoberger et al. ; Yedidi et al.), Lon (Alhuwaider et Dougan ; Bittner et al. ; Fishovitz et al.), p97 (Hänzelmann et Schindelin ; Saffert et al. ; Ye et al.), Pex1/6 (Saffert et al.), CbbQ (Mueller-Cajar), rubisco activase (Bhat et al.), torsines (Chase et al.), et protéases AAA+ mitochondriales (Glynn). Nous présentons ici ces travaux fascinants.
Etudes sur ClpXP, Lon, et les protéases ATP-dépendantes apparentées
Dans leur article de recherche, « The Protein Chaperone ClpX Targets Native and Non-native Aggregated Substrates for Remodeling, Disassembly, and Degradation with ClpP », LaBreck et al. réalisent une série d’expériences élégantes pour établir que ClpX possède une activité de désagrégation contre les polypeptides qui contiennent des signaux spécifiques de reconnaissance de ClpX (LaBreck et al.). En présence de ClpP, ClpX couple la désagrégation de ces substrats à leur dégradation. De manière importante, ils établissent également que ClpXP empêche l’accumulation d’agrégats formés par des protéines portant des signaux de reconnaissance ClpX in vivo (LaBreck et al.). Ces études éclairent le rôle de ClpX en tant que désagrégase de protéines, qui était auparavant sous-estimé.
Dans leur article de recherche, » The Essential Role of ClpXP in Caulobacter crescentus Requires Species Constrained Substrate Specificity « , Vass et al. explorent les fonctions spécifiques aux espèces de ClpX (Vass et al.). Curieusement, ClpX est essentiel chez certaines espèces comme C. crescentus, mais pas chez d’autres bactéries comme E. coli (Vass et al.). Il est important de noter que la ClpX d’E. coli était incapable de compléter la ClpX de C. crescentus in vivo (Vass et al.). Ce manque d’activité était dû à des différences spécifiques à l’espèce dans le domaine N-terminal de ClpX, qui sont critiques pour le traitement de la sous-unité de chargement du clamp de réplication DnaX chez C. crescentus. Ainsi, de petites différences dans la spécificité de ClpX peuvent être particulièrement critiques pour des espèces bactériennes spécifiques.
Dans leur revue sur la « diversité fonctionnelle des complexes protéases AAA+ chez Bacillus subtilis », Elsholz et al. discutent des fonctions de plusieurs protéases AAA+ chez B. subtilis, à savoir : ClpCP, ClpEP, ClpXP, ClpYQ, LonA/B, et FtsH (Elsholz et al.). Ils discutent de la manière dont différentes réponses au stress contrôlent leur expression et des phénotypes observés lors de la délétion de ces différentes protéases. Ils décrivent la capacité de certaines de ces protéases à contrôler la compétence, la sporulation, la motilité et la formation de biofilms. Enfin, les auteurs discutent du ciblage de ces protéases pour le développement de nouveaux antibiotiques.
Dans leur revue intitulée « AAA+ Machines of Protein Destruction in Mycobacteria », Alhuwaider et al. (Alhuwaider et Dougan) discutent des avancées récentes dans la détermination de la structure et de la fonction des protéases AAA+ des mycobactéries. Ces protéases sont : ClpXP1P2, ClpC1P1P2, Lon, FtsH, et Mpa. Les auteurs abordent également le système Pup-protéasome (PPS) présent chez les mycobactéries, qui est équivalent au système ubiquitine-protéasome chez les eucaryotes. Alhuwaider et al. concluent ensuite par une discussion sur les nouveaux composés qui dérégulent ou inhibent l’activité de ClpP1P2 et d’autres qui dérégulent ClpC1. Ces composés ont des activités prometteuses contre les mycobactéries.
Dans un article de recherche intitulé « The Copper Efflux Regulator CueR Is Subject to ATP-Dependent Proteolysis in Escherichia coli », Bittner et al. démontrent que les protéases AAA+ Lon, ClpXP, et ClpAP sont responsables de la dégradation de E. coli CueR, qui est un facteur de transcription qui contrôle l’induction du système Cue d’efflux de cuivre (Bittner et al.). Les auteurs ont découvert que la reconnaissance de CueR par les protéases AAA+ nécessite l’accès à l’extrémité C-terminale de CueR. Ils concluent que les protéases ATP-dépendantes sont nécessaires à l’homéostasie du cuivre chez E. coli.
Fishovitz et al. effectuent une comparaison détaillée entre Lon humaine et E. coli dans leur article de recherche intitulé « Utilization of Mechanistic Enzymology to Evaluate the Significance of ADP Binding to Human Lon Protease » (Fishovitz et al.). En utilisant une étude mécaniste détaillée, ils ont découvert que, contrairement à la Lon d’E. coli, la Lon humaine a une faible affinité pour l’ADP bien qu’elle présente des valeurs de kcat et de KM comparables dans l’activité ATPase. Ils proposent que la Lon humaine ne soit pas régulée par un mécanisme d’échange ADP/ATP favorisé par le substrat. Ces différences entre la Lon humaine et la Lon d’E. coli pourraient permettre le développement futur d’inhibiteurs de la Lon spécifiques à l’espèce.
Dans sa revue sur les « Protéases AAA mitochondriales multifonctionnelles », le Dr Glynn discute des deux protéases AAA mitochondriales, i-AAA et m-AAA (Glynn). Toutes deux sont des protéines de la membrane interne des mitochondries. Cependant, i-AAA projette les domaines ATPase et protéase dans l’espace intermembranaire mitochondrial, tandis que la protéase m-AAA projette les domaines catalytiques dans la matrice. Les structures de ces protéases sont discutées ainsi que leur mécanisme de fonctionnement. Les protéases peuvent effectuer une dégradation complète du substrat, mais aussi ne peuvent cliver que certains substrats comme pour MrpL32 et Atg32.
ClpB et Hsp104
Dans leur mini-revue, « Structural Elements Regulating AAA+ Protein Quality Control Machines », Chang et al. discutent de la façon dont la boucle de pore-1, le motif de signalisation inter-sous-unités et le motif d’insertion du pré-capteur I pourraient contribuer à l’activité de deux désagrégations de Hsp100, la ClpB bactérienne et la Hsp104 de levure (Chang et al.). Ils proposent un modèle pour expliquer comment ces éléments structurels pourraient permettre au cycle AAA+ ATPase d’être couplé à la translocation du substrat à travers le canal central de ClpB et Hsp104. On pense que ce processus de translocation des polypeptides sous-tend la façon dont ClpB et Hsp104 extraient les polypeptides des structures agrégées (Chang et al.).
Duran et al. fournissent une » analyse comparative de la structure et de la fonction des moteurs AAA+ ClpA, ClpB et Hsp104 : fils communs et fonctions disparates » dans leur revue (Duran et al.). Ils discutent de la capacité de ces trois protéines AAA+ (ClpA, ClpB, et Hsp104) à transloquer des polypeptides à travers leurs complexes hexamériques. Toutes ces protéines possèdent deux domaines AAA+ et sont connues pour déplier les protéines. Il est important de noter que ClpB et Hsp104 sont également connues pour fonctionner comme des désagrégats, tandis que ClpA peut former un complexe avec la protéase ClpP. Les auteurs soulignent la nécessité d’utiliser des méthodes cinétiques à l’état transitoire pour examiner les mécanismes cinétiques de ces protéines motrices. Ils décrivent comment l’utilisation de telles méthodes leur a permis de montrer que, par exemple, ClpA transloque des polypeptides à environ 20 aa s-1, tandis qu’en complexe avec la protéase ClpP, la vitesse de translocation de ClpA est encore plus élevée, à environ 35 aa s-1. Les auteurs discutent également de l’importance de la chaperonne Hsp70 dans la fonction de ClpB/Hsp104,et de l’observation d’une spécificité d’espèce dans l’interaction entre Hsp70 et ClpB/Hsp104.
Dans leur article de recherche intitulé « Mutant Analysis Reveals Allosteric Regulation of ClpB Disaggregase », Franke et al. réalisent une analyse mutationnelle sur la ClpB disaggregase d’E. coli pour caractériser sa régulation allostérique (Franke et al.). La ClpB peut être divisée en un domaine N-terminal et deux domaines AAA+ séparés par une région hélicoïdale appelée le domaine M. Les auteurs identifient un résidu hautement conservé dans le domaine M de la ClpB. Les auteurs identifient un résidu hautement conservé dans le premier domaine AAA+, A328. Le mutant ClpB-A328V s’est avéré avoir une activité ATPase très élevée et a présenté une toxicité cellulaire. De manière inattendue, l’activité ATPase élevée de ClpB-A328V était principalement due au deuxième anneau AAA+, comme l’a évalué la spectrométrie de masse par échange d’hydrogène amide. Les auteurs concluent que A328 est un résidu crucial dans le contrôle de l’hydrolyse de l’ATP dans les deux anneaux AAA+ de ClpB.
Dans leur article de recherche intitulé « Substrate Discrimination by ClpB and Hsp104 », Johnston et al. décrivent les préférences de substrat innées de ClpB et Hsp104 en l’absence des systèmes chaperons DnaK et Hsp70 (Johnston et al.). Ils montrent que la spécificité du substrat est déterminée par le premier domaine AAA+ de chaque protéine. Ils sont parvenus à cette conclusion en testant la capacité des deux chaperons à agir sur plusieurs substrats modèles. Ils ont également testé différentes chimères des deux chaperons.
Dans « Hsp78 (78 kDa Heat Shock Protein), a Representative AAA Family Member Found in the Mitochondrial Matrix of Saccharomyces cerevisiae », Abrahão et al. discutent de la structure et de la fonction de Hsp78 (Abrahao et al.). Hsp78 est le paralogue mitochondrial de Hsp104, qui fonctionne dans la désagrégation et la réactivation des protéines (Abrahao et al.). Curieusement, Hsp104 et Hsp78 ont été perdues lors de la transition des protozoaires aux métazoaires (Abrahao et al.). Cependant, Abrahao et al. évoquent l’existence de l’ANKCLP, qui apparaît aux côtés de Hsp78 et Hsp104 chez les protozoaires et survit à la transition évolutive vers les métazoaires. L’ANKCLP possède un domaine AAA+ similaire au domaine 2 de liaison aux nucléotides (NBD2) de Hsp104 et Hsp78, mais est autrement très divergent. De manière intrigante, les mutations de l’ANKCLP provoquent une acidurie 3-méthylglutaconique, une atrophie progressive du cerveau, une déficience intellectuelle, une neutropénie congénitale, des cataractes et des troubles du mouvement chez l’homme (Abrahao et al.).
p97
Dans leur revue, « Structure and Function of p97 and Pex1/6 Type II AAA+ Complexes », Saffert et al. discutent de deux complexes AAA+ différents qui remodèlent les protéines substrats ubiquitinées (Saffert et al.). L’une des fonctions de p97 est de déplacer les substrats ubiquitinés de la membrane du RE vers le protéasome pendant la dégradation associée au RE (Saffert et al.). En revanche, Pex1/Pex6 est un moteur hétérohexamérique composé de sous-unités Pex1 et Pex6 alternées, qui est essentiel pour la biogenèse et la fonction des peroxysomes. Les structures récentes de p97 et de Pex1/6 obtenues par cryomicroscopie électronique (cryomicroscopie) sont discutées et les différences structurelles clés sont mises en évidence.
Dans leur revue intitulée « A Mighty ‘Protein Extractor’ of the Cell : Structure and Function of the p97/CDC48 ATPase », Ye et al. résument les connaissances actuelles sur la structure et la fonction de p97 et son rôle dans plusieurs maladies (Ye et al.). p97 possède deux domaines AAA+ reliés par un court linker. Elle possède également un domaine N-terminal, qui sert de médiateur à ses interactions avec différentes protéines adaptatrices. Les auteurs présentent une discussion détaillée de la structure de la p97 et de l’effet des nucléotides sur ses différentes conformations. Ces études sont basées sur l’utilisation de techniques telles que la ME, la cristallographie aux rayons X et la microscopie à force atomique à haute vitesse. Les auteurs discutent ensuite des fonctions multicellulaires de cette protéine hautement conservée, y compris ses rôles dans la dégradation des protéines associée au RE (ERAD), la dégradation associée à la mitochondrie (MAD) en extrayant les polypeptides de la membrane externe mitochondriale, et la dégradation associée au ribosome (RAD). Enfin, Ye et al. fournissent un résumé des mutations de p97 conduisant à plusieurs maladies humaines telles que l’IBMPFD (Inclusion Body Myopathy associated with Paget’s disease of the bone and Frontotemporal Dementia)], la FALS (sclérose latérale amyotrophique familiale), la CMT2Y (maladie de Charcot-Marie-Tooth, type 2Y), les paraplégies spastiques héréditaires (HSP), la maladie de Parkinson (PD) et la maladie d’Alzheimer (AD).
Dans leur revue sur la p97 intitulée « The Interplay of Cofactor Interactions and Post-translational Modifications in the Regulation of the AAA+ ATPase p97 », Hänzelmann et Schindelin discutent de la façon dont différents cofacteurs modulent l’activité de l’ATPase p97 (Hanzelmann et Schindelin). Ils soulignent le fait que la capacité de la p97 à être impliquée dans un grand nombre de processus cellulaires est due au grand nombre de cofacteurs qui interagissent avec cette protéine. Ils élucident trois classes différentes de cofacteurs de p97, à savoir : (i) les cofacteurs de recrutement de substrat comme les protéines UBA-UBX et UFD1-NPL4, (ii) les cofacteurs de transformation de substrat comme les ubiquitine (E3) ligases et les déubiquitinases (DUB), et (iii) les cofacteurs de régulation comme les protéines UBX, qui peuvent séquestrer ou recycler les hexamères de p97. Les auteurs discutent également du rôle des modifications post-traductionnelles sur l’activité de p97, et sur ses interactions avec ses cofacteurs et substrats.
Protéines AAA+ du protéasome
Dans « AAA-ATPases in Protein Degradation », Yedidi et al. passent en revue les activités de Rpt1, Rpt2, Rpt3, Rpt4, Rpt5 et Rpt6, qui sont les ATPases AAA+ du protéasome eucaryote, ainsi que certaines de leurs parentes bactériennes comme PAN, Mpa et VAT (Yedidi et al.). Ils se concentrent sur les nouvelles technologies pour comprendre comment ces AAA+ ATPases fonctionnent en transloquant les polypeptides dépliés dans la chambre protéolytique de la protéase (Yedidi et al.). Les changements de conformation au sein de l’anneau AAA+ et de la protéase à chambre adjacente semblent générer un mécanisme de pompage péristaltique pour délivrer les substrats à dégrader (Yedidi et al.).
Dans leur article de recherche, « The Proteasomal ATPases Use a Slow but Highly Processive Strategy to Unfold Proteins », Snoberger et al. établissent que les protéines protéasomales AAA+ emploient un mécanisme moteur à faible vitesse mais hautement processif pour délivrer les substrats à la cavité protéolytique du protéasome (Snoberger et al.). Ce mécanisme contraste avec celui de ClpX, qui utilise un mécanisme moteur à grande vitesse mais moins progressif pour délivrer les substrats à la protéase ClpP pour la dégradation. Ces différences dans le mécanisme moteur peuvent avoir évolué en réponse à des demandes différentes de leur clientèle spécifique.
Rubisco Activases
Dans leur revue sur « Rubisco Activases : AAA+ Chaperones Adapted to Enzyme Repair » Bhat et al. discutent de la fonction unique de la Rubisco activase (Rca) dans le remodelage de la Rubisco (Bhat et al.). Rca est une chaperonne AAA+ qui est hautement conservée dans les organismes photosynthétiques, des bactéries aux plantes supérieures. La Rubisco est une enzyme Ribulose-1,5-bisphosphate carboxylase/oxygénase, qui est impliquée dans la fixation du CO2 atmosphérique pendant la photosynthèse. Il s’agit de la protéine la plus abondante sur terre et de l’enzyme clé dans la synthèse de toute la matière organique de la planète. Cependant, la Rubisco est une enzyme pauvre et est facilement inhibée par les produits secondaires de ses réactions catalytiques ou par des composés synthétisés par certaines plantes dans des conditions de faible luminosité. Rca a pour fonction d’atténuer ou de » guérir » la Rubisco de ces inhibitions problématiques. Les auteurs discutent de la structure de Rca de différentes espèces et des mécanismes potentiels de sa fonction.
Le Dr Mueller-Cajar fournit une revue sur « The Diverse AAA+ Machines that Repair Inhibited Rubisco Active Sites » (Mueller-Cajar). Il discute de la présence de trois classes évolutivement distinctes d’activases de Rubisco (Rcas) : (1) les Rcas de type vert et (2) de type rouge que l’on trouve principalement chez les eucaryotes photosynthétiques de la lignée des plastides verts et rouges, respectivement, et (3) les CbbQO présentes chez les bactéries chimioautotrophes. Il discute de l’évolution de ces activases et de leur utilisation potentielle en biologie synthétique pour améliorer l’activité de la Rubisco chez les plantes.
Torsin
Dans leur article de perspective, « Torsin ATPases : Harnessing Dynamic Instability for Function », Chase et al. discutent des Torsines, qui sont également phylogénétiquement liées au NBD2 de la levure Hsp104 (Chase et al.). Les torsines sont les seules ATPases AAA+ localisées à l’intérieur du RE et de l’enveloppe nucléaire connectée (Chase et al.). De manière intrigante, les mutations de TorsinA provoquent la dystonie DYT1, un trouble neurologique chez l’homme (Chase et al.). Les torsines présentent une faible activité ATPase qui est augmentée par la complémentation du site actif due au co-assemblage avec les cofacteurs accessoires spécifiques LAP1 et LULL1 (Chase et al.). Chase et al. suggèrent que l’assemblage et le désassemblage dynamiques des complexes Torsin/cofacteurs jouent des rôles importants dans leur fonction dans le trafic nucléaire et l’assemblage du complexe noyau-pore (Chase et al.).
Pontine et Reptine
Dans leur revue approfondie sur « Le rôle de la Pontine et de la Reptine dans la physiologie cellulaire et l’étiologie du cancer », Mao et Houry discutent des multiples fonctions des ATPases AAA+ hautement conservées de la Pontine et de la Reptine (Mao et Houry). Ces deux protéines fonctionnent généralement ensemble sous la forme d’un complexe, mais peuvent également fonctionner indépendamment. Les auteurs soulignent les rôles de la Pontine et de la Reptine dans le remodelage de la chromatine. Ils discutent également de la manière dont la Pontine et la Reptine modulent les activités transcriptionnelles de plusieurs proto-oncogènes tels que MYC et la β-caténine. Mao et Houry expliquent comment la Pontine et la Reptine sont requises pour l’assemblage des complexes de signalisation PIKK ainsi que pour la télomérase, le fuseau mitotique, l’ARN polymérase II et les snoRNP. Les auteurs concluent par un aperçu des efforts actuels visant à identifier des inhibiteurs de la Pontine et de la Reptine à développer comme nouveaux anticancéreux.
Marques finales
En conclusion, cette collection de 21 articles met en évidence un certain nombre d’aspects structurels et mécanistiques importants des protéines AAA+ impliquées dans la réparation et la dégradation des protéines. Nous sommes enthousiastes à l’idée de voir comment le domaine va continuer à se développer au cours de la révolution en cours de la cryo-EM (Egelman, 2016). Nous prévoyons que la cryo-EM permettra une compréhension plus profonde de la façon dont ces fascinantes machines moléculaires fonctionnent dans diverses situations (Gates et al., 2017 ; Puchades et al., 2017 ; Ripstein et al., 2017 ; Zehr et al…, 2017).
Contributions des auteurs
Tous les auteurs listés ont apporté une contribution substantielle, directe et intellectuelle à ce travail, et l’ont approuvé pour publication.
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière qui pourrait être interprétée comme un conflit d’intérêts potentiel.
Remerciements
Le travail dans le laboratoire de WH dans ce domaine est financé par une subvention de projet de recherche des Instituts de santé du Canada (PJT-148564). JS est soutenu par la subvention NIH R01GM099836.
Egelman, E. H. (2016). La révolution actuelle dans la cryo-EM. Biophys. J. 110, 1008-1012. doi : 10.1016/j.bpj.2016.02.001
PubMed Abstract | CrossRef Full Text | Google Scholar
Erzberger, J. P., et Berger, J. M. (2006). Relations évolutives et mécanismes structurels des protéines AAA+. Annu. Rev. Biophys. Biomol. Struct. 35, 93-114. doi : 10.1146/annurev.biophys.35.040405.101933
PubMed Abstract | CrossRef Full Text | Google Scholar
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro en 2017-au-delà des annotations de familles et de domaines de protéines. Nucleic Acids Res. 45, D190-D199. doi : 10.1093/nar/gkw1107
PubMed Abstract | CrossRef Full Text | Google Scholar
Gates, S. N., Yokom, A. L., Lin, J., Jackrel, M. E., Rizo, A. N., Kendsersky, N. M., et al. (2017). Mécanisme de translocation des polypeptides en forme de cliquet de la désagrégation de l’AAA+ Hsp104. Science 357, 273-279. doi : 10.1126/science.aan1052
PubMed Abstract | CrossRef Full Text | Google Scholar
Hanson, P. I., et Whiteheart, S. W. (2005). Protéines AAA+ : have engine, will work. Nat. Rev. Mol.Cell Biol. 6, 519-529. doi : 10.1038/nrm1684
PubMed Abstract | CrossRef Full Text | Google Scholar
Olivares, A. O., Baker, T. A., et Sauer, R. T. (2016). Aperçus mécanistiques sur les protéases AAA+ bactériennes et les machines à remodeler les protéines. Nat. Rev. Microbiol. 14, 33-44. doi : 10.1038/nrmicro.2015.4
PubMed Abstract | CrossRef Full Text | Google Scholar
Puchades, C., Rampello, A. J., Shin, M., Giuliano, C. J., Wiseman, R. L., Glynn, S. E., et al. (2017). La structure de la protéase AAA+ de la membrane interne mitochondriale YME1 donne un aperçu du traitement du substrat. Science 358:eaao0464. doi : 10.1126/science.aao0464
PubMed Abstract | CrossRef Full Text | Google Scholar
Ripstein, Z. A., Huang, R., Augustyniak, R., Kay, L. E., et Rubinstein, J. L. (2017). Structure d’une unfoldase AAA+ dans le processus de dépliage du substrat. Elife 6:e25754. doi : 10.7554/eLife.25754
PubMed Abstract | CrossRef Full Text | Google Scholar
Snider, J., Thibault, G., et Houry, W. A. (2008). La superfamille AAA+ de protéines fonctionnellement diverses. Genome Biol. 9:216. doi : 10.1186/gb-2008-9-4-216
PubMed Abstract | CrossRef Full Text | Google Scholar
Zehr, E., Szyk, A., Piszczek, G., Szczesna, E., Zuo, X., et Roll-Mecak, A. (2017). Les structures en spirale et en anneau de la Katanine éclairent le coup de force pour la section des microtubules. Nat. Struct. Mol. Biol. 24, 717-725. doi : 10.1038/nsmb.3448
PubMed Abstract | CrossRef Full Text | Google Scholar
.