SONAL DESAI, ARCHITA PATEL ET S. Y. GABHE*
C. U. Shah College of Pharmacy, S. N. D. T. Women’s University, Sir Vithaldas Vidya Vihar, Juhu Tara Road, Santacruz (W), Mumbai – 400 049, India
Auteur correspondant : S. Y. GABHE Courriel :
| Date d’acceptation | 16-Jan-2011 |
| Date de révision | 26-.Oct-2010 |
| Date de réception | 9-Avr-2009 |
DOI : 10.4103/0250-474X.89762
Abstract
Une simple chromatographie liquide haute performance isocratique en phase inverse a été utilisée pour séparer trois impuretés présentes dans l’échantillon de 8-chlorotheophylline. La LC-MS a été utilisée pour la caractérisation des impuretés. Sur la base des données de spectres de masse, les structures de ces impuretés ont été caractérisées comme étant la 3,7-dihydro-1,3-diméthyl-1H-purine-2,6-dione (impureté I), la 3,7-dihydro-1,3,7-triméthyl-1H-purine-2,6-dione (impureté II) et l’isomère de la 8-chloro- 1,3-diméthyl-2,6(3H,1H)-purinedione (impureté III).
Mots-clés
8-Chlorotheophylline, impureté, LC-MS, HPLC en phase inversée, théophylline
Introduction
L’industrie des médicaments en vrac constitue la base de toutes les industries pharmaceutiques, car elle est la source d’ingrédients pharmaceutiques actifs (API) de qualité spécifiée. Le défi majeur pour les industries de médicaments en vrac est de produire le médicament final de qualité et de pureté requises, de manière économique. La pureté des IPA dépend de plusieurs facteurs tels que les matières premières, leurs méthodes de fabrication et le type de processus de cristallisation ou de purification. Cependant, il est presque impossible d’obtenir des matières absolument pures, car des impuretés s’y incorporent soit pendant la fabrication, la purification ou le stockage. Une impureté est tout composant d’une substance médicamenteuse (à l’exclusion de l’eau) qui n’est pas une entité chimique définie comme substance médicamenteuse. Pour la substance médicamenteuse produite par synthèse chimique, l’ICH classe les impuretés en trois catégories : les impuretés organiques, les impuretés inorganiques et les solvants résiduels. Les impuretés présentes dans la substance peuvent être toxiques, modifier les propriétés physiques ou chimiques de la substance, la rendant ainsi médicalement inutile. Les impuretés peuvent réduire la durée de conservation du produit et causer des difficultés dans la formulation. Par conséquent, non seulement le contrôle des impuretés mais aussi la qualification des impuretés est une question critique pour les industries de médicaments en vrac.
Les antihistaminiques comme le dimenhydrinate et la prométhazine théoclate sont des médicaments largement utilisés dans le traitement du mal des transports. La 8-chlorotheophylline, chimiquement 8-chloro-1,3-dimethyl-2,6(1H, 3H)- purinedione, est un intermédiaire qui est utilisé dans la préparation de la forme sel de ces médicaments. Il est essentiel de garantir la pureté et la sécurité du dimenhydrinate et du théoclate de prométhazine. Pour cela, la 8-chlorotheophylline doit être obtenue avec la plus grande pureté et avec un profil d’impureté connu.
D’après l’étude de la littérature, on constate que Wadke et al. ont étudié les interactions de la 9-méthylisoalloxazine et de la 3,9-diméthylisoalloxazine avec la 8-chlorotheophylline. La 8-chlorotheophylline a été utilisée comme standard interne pour la détermination de l’urate par la méthode HPLC. Elle a également été déterminée par potentiométrie. La détermination simultanée du chlorhydrate de chlorphénoxamine, de la 8-chlorotheophylline et de la caféine dans une forme posologique à plusieurs composants a été réalisée par une méthode de chromatographie en couche mince-densitométrique. Gil et al. ont étudié le comportement électroanalytique de la 8-chlorotheophylline par voltammétrie cyclique et par polarographie différentielle à impulsions et ont déterminé sa teneur dans des préparations pharmaceutiques par polarographie différentielle à impulsions. Une méthode RP-HPLC indiquant la stabilité a été développée et validée pour la 8-chlorotheophylline avec la diphenhydramine et la caféine. Des méthodes spectrophotométriques et chimiométriques de premier ordre à passage par zéro ont également été développées pour la détermination simultanée de la caféine, de la 8-chlorotheophylline et du chlorhydrate de chlorphénoxamine dans des mélanges ternaires. Jusqu’à présent, aucune méthode chromatographique et spectroscopique n’a été rapportée pour la séparation et la caractérisation des impuretés présentes dans la 8-chlorotheophylline. Par conséquent, le présent travail a été entrepris dans le but d’isoler et de caractériser les impuretés présentes dans la 8-chlorotheophylline en utilisant des techniques analytiques modernes.
La 8-chlorotheophylline était un échantillon cadeau de Kores (India) Ltd, Thane. Tous les autres produits chimiques et réactifs ont été obtenus auprès de S. D. Fine Chemicals Ltd (Mumbai, Inde). Les solvants utilisés pour les études TLC et TLC préparative étaient de qualité analytique et ceux utilisés pour les études HPLC étaient de qualité HPLC. L’acétate de sodium trihydraté de qualité AR a été utilisé pour la préparation du tampon.
Initialement, des études TLC ont été réalisées afin de connaître le nombre d’impuretés présentes dans l’échantillon. Des plaques CCM pré-revêtues de gel de silice 60GF254 (Merck) ont été utilisées comme phase stationnaire. L’échantillon a été dissous dans une quantité minimale d’acétate d’éthyle et cette solution a été utilisée pour le marquage des plaques CCM. Diverses phases mobiles ont été essayées. L’acétate d’éthyle:toluène:acide acétique glacial (10:0,3:0,5 v/v/v) a montré une meilleure séparation par rapport aux autres phases mobiles. Quatre composants ont été séparés de l’échantillon de 8-chlorotheophylline en utilisant la CCM avec les valeurs Rf de 0,029, 0,132, 0,198 et 0,852, respectivement. La 8-chlorotheophylline avait un Rf de 0,852.
Une fois la phase mobile pour la CCM développée, on a essayé de séparer le mélange en utilisant la CCM préparative. L’échantillon a été dissous dans une quantité minimale d’acétate d’éthyle et repéré sous forme de bande. Les trois impuretés ont été séparées de la 8-chlorotheophylline par CCM préparative en utilisant les mêmes conditions chromatographiques que celles utilisées dans les études CCM. Les différentes bandes ont été recueillies séparément et extraites à l’aide d’acétate d’éthyle. Comme les quantités de chacune des impuretés I, II et III isolées étaient très faibles, il a été décidé d’isoler ces impuretés collectivement. Les impuretés I, II et III ont été désignées collectivement comme la fraction I. La fraction I a été isolée de la 8-chlorothéophylline par CCM préparative. Comme chaque impureté n’a pas été isolée séparément, différentes techniques d’identification telles que l’IR, la RMN n’ont pas pu être entreprises. Il a été décidé de procéder à une étude plus approfondie en utilisant la LC-MS, qui permet une séparation et une caractérisation simultanées. Avant l’analyse LC-MS, le profil HPLC de la fraction I a été développé. Un chromatographe liquide à haute performance Tosoh équipé d’une pompe alternative CCPM, d’un contrôleur de pompe PX8010 et d’un détecteur UV a été utilisé pour les études HPLC. Une boucle d’une capacité de 20 µl a été fixée à l’unité de vanne d’injection. La fraction I a été dissoute dans de l’acétonitrile et a été soumise à une analyse HPLC en phase inversée avec une phase mobile comprenant de l’acétonitrile : acétate de sodium trihydraté (pH 3,57 ; 0,01 M) (5:95 v/v). La colonne choisie était la colonne Phenomenex ODS (250×4,6 mm I.D. ; taille des particules 5 µm). Le débit était de 1,5 ml/min et la détection était contrôlée à une longueur d’onde de 280 nm. La phase mobile a été filtrée à travers un filtre en verre fritté G5 sous vide avant utilisation et soniquée pour éliminer les bulles d’air. L’analyse HPLC de la fraction I a également révélé trois pics avec les temps de rétention moyens de 8,324 min, 14,275 min et 20,933 min, respectivement (fig. 1). Le pic avec un temps de rétention moyen de 2,086 min était le pic du solvant.
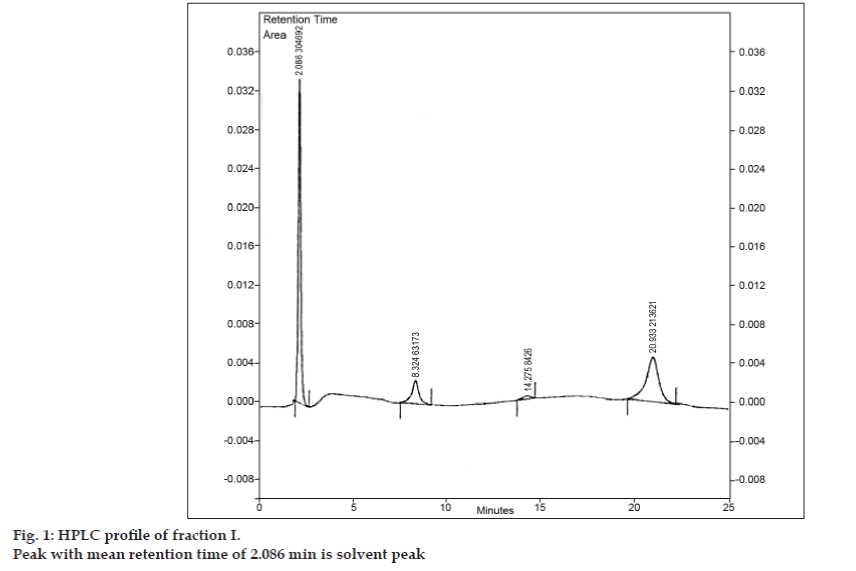
Figure 1 : profil HPLC de la fraction I.
La fraction I a ensuite été soumise à une analyse LC-MS pour la caractérisation des impuretés. Les études LC-MS ont été réalisées sur un système dans lequel la partie LC était constituée d’une HPLC de la série 1100 (Agilent Technologies, USA) comprenant un dégazeur sous vide (G1322A), une pompe quaternaire (G1311A), un auto-échantillonneur (G1313A) et un détecteur UV/visible (G1314A) et la partie MS était constituée d’un spectromètre de masse triple quadripolaire Quattro II (Micromass UK Ltd, UK).
Dans les études LC-MS, les séparations par chromatographie liquide ont été réalisées par une colonne Phenomenex C18 (250×4,6 mm, 5 µm) à température ambiante avec la phase mobile d’acétonitrile : tampon acétate de sodium (5:95, v/v) à un débit de 1,5 ml/min. Le spectromètre de masse a fonctionné en mode d’ionisation par pulvérisation d’électrons positifs (ESI) avec un rapport masse/charge (m/z) dans la plage de 120-500 m/z. De l’azote a été utilisé comme gaz de nébulisation. Les données ont été acquises et traitées par le logiciel Masslynx. Les données du spectre de masse des impuretés ont été obtenues (figures 2, 3 et 4). La voie de fragmentation pour trois pics est caractérisée par la perte du groupe méthyle et/ou du groupe carbonyle. Les pics avec des valeurs m/z de 180,9, 195,7 et 214,9 correspondent respectivement à des pics +, + et +. Selon les données MS obtenues, l’impureté éluant à 8,324 min, 14,275 min et 20,933 min était la théophylline (poids molaire 180), la caféine (poids molaire 194) et un isomère de la 8-chlorotheophylline (poids molaire 214,5), respectivement (Tableau 1). Ainsi, trois impuretés ont été séparées et leurs structures ont été élucidées sur la base des données de spectres de masse (Schéma 1).
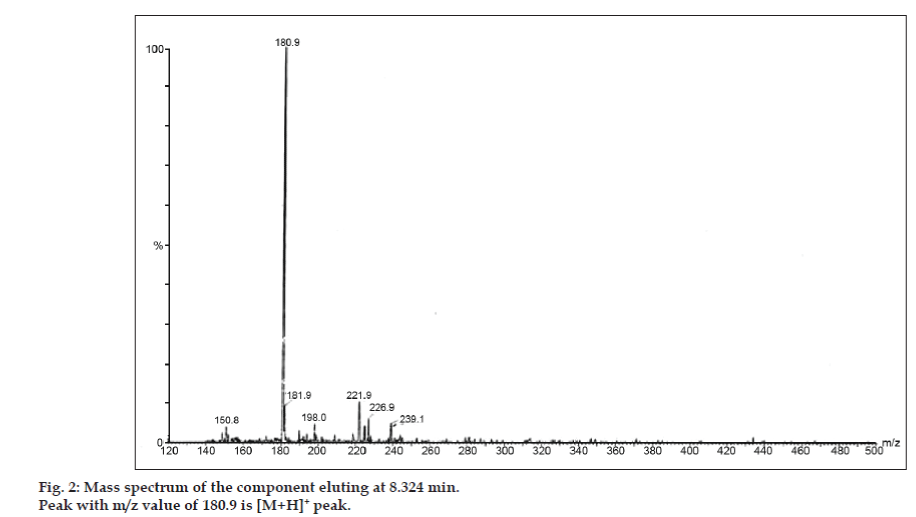
Fig 2 : Spectre de masse du composant éluant à 8,324 min.
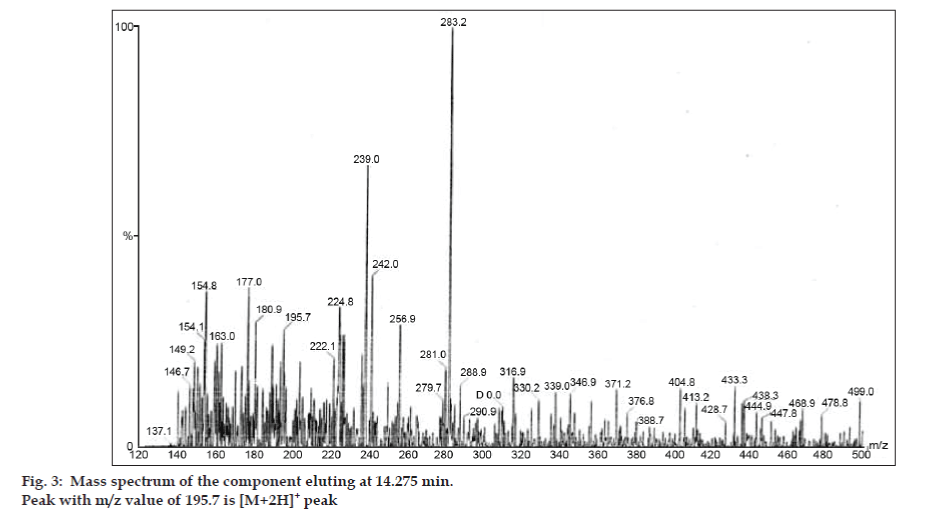
Fig. 3 : Spectre de masse du composant éluant à 14,275 min.
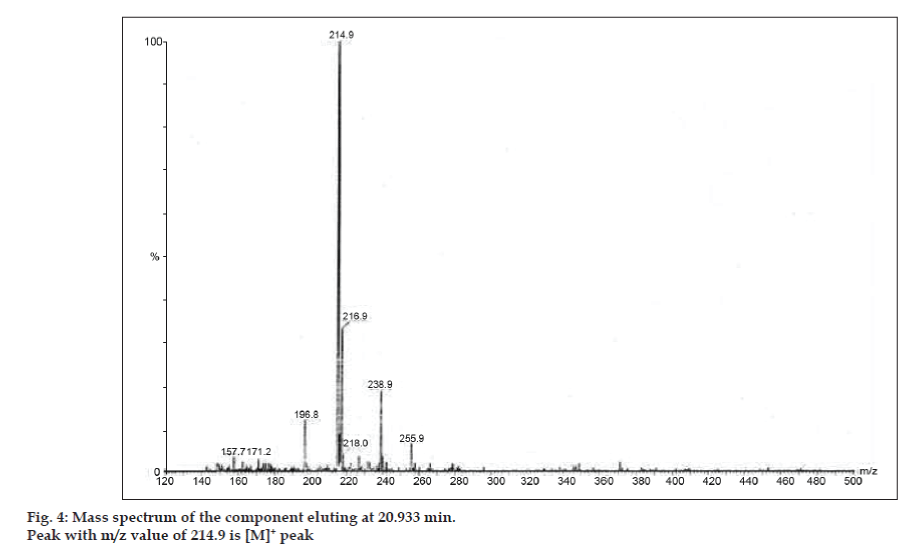
Fig. 4 : Spectre de masse du composant éluant à 20,933 min.
| Peak no. |
Temps de rétention (min) |
Ions fragments (m/z) | Identification |
|---|---|---|---|
| 8.324 | 181,9 +, 180,9 +, 150,8 + | Théophylline | |
| 14.275 | 195,7 +, 180,9 +,149,2 +, 137,1+ | Caféine | |
| 20,933 | 216.9 +, 214,9 +, 171,2 +, 157,7 + | Isomère de la 8- Chlorothéophylline |
TABLEAU 1 : IDENTIFICATION HPLC-MS DE LA FRACTION I
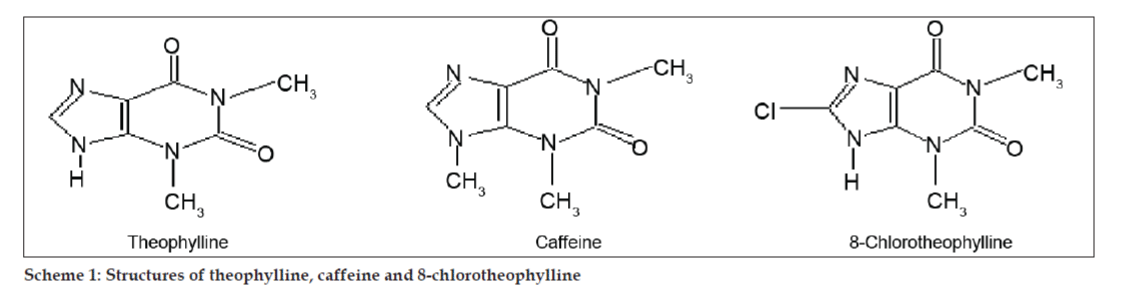
Schéma 1 : Structures de la théophylline, de la caféine et de la 8-chlorotheophylline
Reconnaissance
Les auteurs remercient Kores (India) Ltd, Thane pour avoir fourni un échantillon cadeau de 8-chlorotheophylline.
- Kasture AV, Wadodkar SG, Mahadik KR, More HV. Analyse pharmaceutique. Vol. 1. Pune : NiraliPrakashan ; 1997. p. 12-4.
- Pharmacopée des États-Unis, Vol. 26. Rockville, MD : United States Pharmacopoeia Convention, Inc. ; 1999. p. 2049-59.
- Ahuja S, Alsante KM. Manuel d’isolement et de caractérisation des impuretés dans les produits pharmaceutiques. Californie : Academic Press ; 2003. p. 6.
- Foye WO, Lemke TL, Williams DA. Principes de chimie médicinale. 4th ed. New Delhi : B. I. Waverly Pvt Ltd ; 1995. p. 419.
- Reynolds JE. Martindale-The Extra Pharmacopoeia. 29th ed. Londres : Pharmaceutical Press ; 1989. p. 451,459.
- Wadke DA, Guttman DE. Influence de la formation de complexes sur la vitesse de réaction III. Interaction de certaines isoalloxazines avec la 8-chlorotheophylline comme déterminé par des méthodes spectrales, de solubilité et cinétiques. J Pharm Sci 1965;54:1293.
- Bennett MJ, Patchett BP, Worthy E. Une méthode HPLC simple pour la détermination de l’urate dans le sérum et l’urine en utilisant la 8-chlorotheophylline comme standard interne. Med Lab Sci 1984;41:108-11.
- Nikolic K, Medenica M. Détermination potentiométrique de la 8-chlorotheophylline. Microchimica Acta 1986;88:5.
- Bebawy LI, El-Kousy NM. Détermination simultanée de certaines formes galéniques à plusieurs composants par la méthode densitométrique quantitative de chromatographie sur couche mince. J Pharm Biomed Anal 1999;20:663-70.
- Gil EP, Blazquez LC, Garcia-MoncoCarra RM, Misiego AS. Comportement polarographique de la 8-Chlorotheophylline et sa détermination dans les formes de dosage. Electroanalysis 1993;5:343.
- Barbas C, Garcia A, Saavedra L, Castro M. Optimisation et validation d’une méthode de détermination de la caféine, de la 8-chlorotheophylline et de la diphenhydramine par chromatographie liquide haute performance isocratique Stress test for stability evaluation. J ChromatogrA 2000;870:97-103.
- Kelani KM. Détermination simultanée de la caféine, de la 8- chlorothéophylline et du chlorhydrate de chlorphénoxamine dans des mélanges ternaires par des méthodes spectrophotométriques et chimiométriques à rapport zéro-crossing first-derivative. J AOAC Int 2005;88:1126-34.