Introduction
La polyarthrite rhumatoïde (PR) est une maladie auto-immune, de nature polygénique, caractérisée par une polyarthrite avec des manifestations systémiques et une morbidité accrue et sévère1,2. La PR affecte 0,5 % à 1 % de la population, entraînant une diminution de la qualité de vie, un handicap physique important et un coût économique substantiel.3-6 L’expression clinique de la maladie est variée, allant de formes légères autolimitatives à une évolution très agressive et rapide qui culmine avec la destruction de l’articulation touchée et le handicap qui en résulte7.
Des études génétiques ont confirmé l’existence d’un substrat génétique, en partie lié à certains gènes codant pour des protéines impliquées dans les réponses des cellules T.1 Ces résultats renforcent l’importance du rôle attribué aux cellules T dans l’initiation et la perpétuation de la réponse immunitaire anormale dans cette maladie.8
La pathogénie de la PR est complexe et implique différentes populations cellulaires liées à la réponse immunitaire innée et adaptative. Les cellules résidentes de la synovie, comme les synoviocytes fibroblastiques B ou les macrophages de l’intima, et les cellules inflammatoires du sang comme les lymphocytes T, les lymphocytes B et les monocytes9 sont impliquées dans la pathogenèse. Elles contribuent toutes à la transformation agressive du phénotype des synoviocytes B et au développement d’un infiltrat inflammatoire intense avec pour résultat final la destruction du cartilage et de l’os sous-chondral10,11 (Fig. 1).
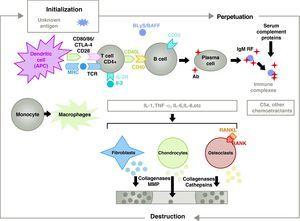
Pathophysiologie de la polyarthrite rhumatoïde. Organisation physiopathologique générale de la polyarthrite rhumatoïde. AC, anticorps ; BAFF, B cell activating factor ; BLyS, B lymphocyte stimulator ; CD, cluster of differentiation ; CPA, antigen-presenting cell ; CPH, MHC ; CTLA4, lymphocyte-associated antigen 4 T cytotoxic C5a fraction complement 5a, FR, facteur rhumatoïde ; Ig, immunoglobuline ; IL, interleukine ; MMP, métalloprotéinases matricielles ; RANK, récepteur activateur du facteur nucléaire B kappa ; RANKL, récepteur activateur ligand du facteur nucléaire B kappa ; RCT, récepteur des cellules T ; TNF, facteur de nécrose tumorale.
Le traitement actuel de la PR repose sur l’administration de médicaments antirhumatismaux modificateurs de la maladie (DMARD) utilisés seuls ou en association12. Ces médicaments ralentissent la destruction des articulations, c’est-à-dire qu’ils sont capables de modifier l’évolution naturelle de la maladie.4,13 Cependant, le pourcentage de patients ayant une réponse clinique satisfaisante est faible et nécessite souvent l’ajout d’un médicament biologique chez un pourcentage élevé de patients.9,13-15
Ces dernières années, de nouvelles molécules et cibles thérapeutiques dont le blocage pourrait réduire ou éliminer la réponse inflammatoire chronique ont été identifiées. L’une de ces nouvelles molécules est l’abatacept. L’abatacept est une construction protéique entièrement humanisée, constituée du domaine extracellulaire de l’antigène 4 associé aux lymphocytes T cytotoxiques humains (CTL4) et d’un fragment génétiquement modifié de la région Fc de l’immunoglobuline G1 humaine (IgG1), qui inhibe les cellules T de costimulation agissant sur le véritable noyau de la réponse immunitaire et, par conséquent, au début de la maladie.
Activation des cellules T
L’activation immunitaire efficace des cellules T nécessite la participation de deux groupes de récepteurs membranaires sur les cellules présentatrices d’antigènes (CPA)14 (figures 1 et 2). Le premier est le véhicule utilisé par les CPA pour fournir l’antigène spécifique préalablement traité à la cellule T. Malgré l’énorme effort consacré à cette recherche, nous ne sommes toujours pas en mesure d’identifier les antigènes arthritogènes qui déclenchent la PR8. La présentation par les CPA d’un antigène contre lequel une réponse immunitaire spécifique est montée est organisée par un complexe trimoléculaire comprenant : des molécules du complexe majeur d’histocompatibilité (CMH) présentes dans la CPA, l’antigène contre lequel la réponse immunitaire se développe et un récepteur membranaire sur la cellule T (TCR) spécifique de cet antigène15 (signal ou voie de signalisation de la réponse immunitaire 1).
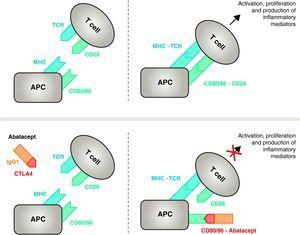
Mécanisme d’action de l’abatacept. Le fragment d’abatacept comprenant le domaine extracellulaire de CTLA4 se lie aux récepteurs CD80/CD86, empêchant ou déplaçant son interaction avec le récepteur CD28. De cette façon, il bloque sélectivement la liaison spécifique de CD80/CD86 au récepteur CD28, ce qui constitue, sur le plan physiopathologique, un blocage du second signal d’activation immunitaire et, par conséquent, de l’activation des cellules T CPA, cellule présentatrice d’antigène ; CMH, complexe majeur d’histocompatibilité ; TCR, récepteur des cellules T.
Pour inhiber l’activation complète, les cellules T ont besoin d’un deuxième ensemble de récepteurs de communication intercellulaire entre les APC et les cellules T qui se produit par le biais des voies de costimulation et constitue la réponse immunitaire dite à 2 signaux14. Bien qu’il existe plusieurs voies de costimulation, l’une d’entre elles est essentielle, à savoir la liaison des récepteurs CD80 (B7-1)/CD86 (B7-2) sur la membrane de l’APC avec le récepteur CD28 sur les cellules T.10,16 L’activation simultanée des deux déclenche une intense signalisation intracellulaire dans les cellules T, essentielle à la pleine activation, à la prolifération, à la survie et à la production de cytokines 8. A 24-48h de l’activation des lymphocytes T, la même signalisation intracellulaire initie un mécanisme de régulation qui vise à désactiver la réponse elle-même. Cela induit l’expression de CTLA411 sur la membrane cellulaire du lymphocyte avec la tâche de concurrencer le CD28 en raison de sa plus grande affinité de liaison avec CD80/CD86.17,18
L’activation des deux sous-ensembles de cellules T, CD4+ et CD8+ dépend du récepteur costimulateur CD28. Les cellules T CD4+ sont des cellules T auxiliaires. Ils reconnaissent les peptides présentés par les molécules du CMH de classe II présentes sur l’APC. Ces antigènes proviennent de la voie exogène qui traite les agents pathogènes tels que les bactéries. De nombreuses maladies auto-immunes sont associées à une réponse pathologique des cellules T CD4+. De leur côté, les cellules T CD8+ sont des lymphocytes cytotoxiques (CTL). Les cellules T CD8+ reconnaissent les antigènes, principalement viraux et tumoraux présentés par les molécules de classe I du CMH. Après activation, les cellules CD8+ assurent la destruction des cellules cibles par la production de perforine, de granzymes et d’interféron (IFN)-g. Les deux sous-types de cellules T sont activés par costimulation avec CD2815, bien que l’activation des cellules T CD8+ soit moins dépendante de cette voie de costimulation. En fait, alors que toutes les cellules CD4+ expriment le CD28 sur leur membrane, ce n’est le cas que d’environ 50 % des cellules CD8+.19 De plus, il a été démontré que les cellules CD4+ présentent une réponse plus importante à la liaison du CD2820. De plus, le promoteur CD28 n’est pas une condition absolue pour l’activation des CTL.21 Tous ces éléments apporteraient un double avantage thérapeutique dans la pratique clinique. D’une part, l’abatacept agit préférentiellement sur la cellule cible dans la pathogenèse de la maladie. D’autre part, l’action réduite sur l’activité des lymphocytes CD8+ assurerait un meilleur profil de sécurité en termes de complications virales et tumorales.
L’activation des cellules T CD4+ est le point de départ d’une cascade pro-inflammatoire avec production de grandes quantités de cytokines et prolifération cellulaire qui, si elle est perpétuée et maintenue, comme dans la PR, conduit à une inflammation chronique très active, capable de détruire les tissus dans lesquels elle se déclenche, principalement les articulations dans le cas de la PR8 (Fig. 1). La synoviale commence à proliférer en raison de l’infiltration de cellules provenant du sang, notamment les lymphocytes T eux-mêmes et leurs sous-types, ainsi que les lymphocytes B. Les monocytes se différencient en macrophages et en ostéoclastes et activent également les chondrocytes articulaires. Dans cet environnement, il y a de grandes quantités de cytokines pro-inflammatoires comme l’interleukine (IL)-1, l’IL-6 et le facteur de nécrose tumorale (TNF) et bien d’autres. Les cellules B produisent également des auto-anticorps tels que le facteur rhumatoïde ou les anticorps anti-peptides citrullinés. Tous ces éléments conduisent à la destruction non seulement de la membrane synoviale mais aussi de l’os et du cartilage sous-jacents.22
La biotechnologie dans le traitement de la polyarthrite rhumatoïde
En raison des recherches susmentionnées, la fabrication biotechnologique de diverses molécules visant à bloquer des cibles spécifiques a été développée et commercialisée. La première génération a été caractérisée par l’apparition de médicaments neutralisant le TNF : l’étanercept, l’infliximab et l’adalimumab et l’anakinra, qui inhibent l’action de l’IL-1. Par la suite, de nouvelles molécules sont apparues, comme l’abatacept, pour moduler la costimulation de la réponse immunitaire, le certolizumab et le golimumab pour bloquer le TNF, le rituximab contre le récepteur CD20 des lymphocytes B, et le tocilizumab qui bloque l’IL-6.7,23-26
Malgré l’énorme bond en avant en termes d’efficacité thérapeutique dû à l’introduction de ces médicaments, un pourcentage substantiel de patients, estimé entre 25% et 40%, ne répond pas aux médicaments ou aux produits biologiques actuellement commercialisés ou est affecté par la survenue d’effets indésirables27.-32 La nécessité d’améliorer cette situation reste un encouragement dans la poursuite et le développement de nouvelles molécules visant à réguler différentes cibles thérapeutiques qui pourraient améliorer l’efficacité thérapeutique, comme le cas de l’abatacept, qui module sélectivement l’activation des cellules T33.
L’abatacept est une construction protéique qui est produite par la technologie de l’ADN recombinant dans des cellules ovariennes de hamster.34,35 Cette molécule a été conçue pour interférer avec la régulation des voies de costimulation dans les cellules T, qui jouent un rôle important dans la pathogenèse de diverses maladies auto-immunes, des infections, du rejet d’organes transplantés et de l’immunité tumorale36.
L’abatacept est utilisé en association avec le méthotrexate chez les patients atteints de PR qui ont eu une réponse inadéquate ou une intolérance aux autres DMARD, y compris le méthotrexate (MTX) ou un inhibiteur du TNF-alpha. Dans l’arthrite juvénile idiopathique polyarticulaire, il est indiqué chez les patients âgés de 6 ans ou plus qui ont eu une réponse inadéquate à d’autres DMARD, y compris au moins un médicament neutralisant le TNF35.
Mécanisme d’action de l’abatacept
L’abatacept est un modulateur sélectif du signal costimulatoire CD80/86-CD28, et comme nous l’avons vu précédemment, il est essentiel pour l’activation des cellules T L’abatacept inhibe l’activation des cellules T, en bloquant sélectivement la liaison spécifique du récepteur CD80/CD86 dans l’APC au CD28 sur la cellule T (Fig. 2).22,37 La stratégie pharmacologique vise à inhiber la réponse immunitaire/inflammatoire accélérée, caractéristique de la maladie, et à rétablir l’homéostasie normale du système immunitaire. En fait, la compétition entre le CD28 endogène et le CTLA4 pour la liaison au CD80/86 est le mécanisme physiologique utilisé pour réguler et, le cas échéant, conclure une réponse immunitaire normale. L’abatacept, en bloquant la liaison de CD80/86 à CD28, inhibe la transmission d’un deuxième signal de la réponse immunitaire, qui produit indirectement un signal négatif sur l’activation des cellules T. De plus, l’abatacept a probablement un effet plus important en empêchant la formation d’un signal costimulatoire dans les cellules T, en inactivant celles déjà actives, qui ne sont pas liées au CTLA4
Médicament d’accompagnement
1. Pourquoi l’abatacept est-il inclus dans le groupe des médicaments immunomodulateurs ? Fondamentalement, parce qu’il produit une déplétion cellulaire, notamment des lymphocytes T en raison de l’action pharmacologique exercée en ne bloquant pas sélectivement une cytokine particulière, évitant ainsi la suppression radicale des voies essentielles au bon fonctionnement de la réponse immunitaire.8
2. Comment empêche-t-il la liaison de la région Fc de la molécule à son récepteur ? La région Fc de l’abatacept est génétiquement modifiée, de sorte qu’elle ne se lie pas aux récepteurs CD16 et CD32, et se lie très faiblement au récepteur CD64. Cette conception permet de contourner les réponses cellulaires médiées par le récepteur Fc, comme la cytotoxicité cellulaire dépendante des anticorps (ADCC) et la cytotoxicité dépendante du complément (CDC).18 Ces deux phénomènes sont associés à la lyse cellulaire, avec des effets indésirables potentiels qui peuvent être observés dans les traitements prolongés38. Par conséquent, le fragment modifié d’IgG1 semble être actif, prévenant ainsi les effets indésirables résultant de l’ADCC.39
3. Effet anti-inflammatoire de l’abatacept. L’abatacept réduit de manière significative un grand nombre de médiateurs inflammatoires chez les patients atteints de PR, les ramenant à la normale, un fait démontré dans plusieurs essais cliniques utilisés lors de la recherche du médicament.
Dans une étude de phase II-b, d’un an, contrôlée par placebo, chez des patients atteints de PR et présentant une réponse inadéquate au MTX, des échantillons ont été prélevés et les niveaux sériques de marqueurs sélectionnés ont été mesurés dans les jours précédant la perfusion afin d’étudier l’effet de l’abatacept sur les médiateurs et les cytokines pro-inflammatoires. Un groupe de patients a reçu du MTX et de l’abatacept à raison de 10mg/kg, selon un calendrier régulier. Le groupe témoin, quant à lui, a été traité par MTX et placebo. Un an après le traitement, les marqueurs dans le groupe abatacept de 10mg/kg s’étaient normalisés, alors qu’ils restaient élevés dans le groupe placebo (TNF : 7,4 vs 10,3pg/ml ; FR : 159 vs 225U/l, sIL-2R : vs 1228,3. 1697,1pg/ml IL-6 : 7,3 vs 19,9pg/ml).40
4. Immunogénicité. Selon les données sur le médicament, seuls 187 des 3877 (4,8%) patients atteints de PR traités jusqu’à 8 ans avec l’abatacept ont développé des anticorps contre le médicament pendant le traitement.41 Les anticorps contre l’abatacept ont été évalués chez les patients après l’arrêt du médicament (>42 jours après la dernière dose), et chez 103 des 1888 (5,5%) étaient séropositifs. En revanche, dans une autre étude portant sur 2000 patients abatacept, les anticorps ont été mesurés et il a été conclu que l’abatacept a une faible immunogénicité.42,43
5. Abatacept et tuberculose. Le TNF participe à la réponse inflammatoire et à l’immunopathologie de la tuberculose (TB). Des études in vitro montrent que le TNF augmente l’activité phagocytaire et mycobactéricide des macrophages, tandis qu’in vivo, il est impliqué dans la formation initiale et le maintien ultérieur des granulomes, ce qui contrôle la croissance des mycobactéries et limite leur propagation. Dans un modèle chronique de réactivation de la tuberculose latente chez la souris, nous avons étudié l’évolution de l’infection chez des souris traitées par abatacept par rapport à un autre groupe traité par un anti-TNF monoclonal murin.42 Quatre mois après avoir infecté des souris C57BL/6 avec Mycobacterium tuberculosis et, une fois confirmée leur infection tuberculeuse latente, les souris ont été traitées pendant 16 semaines avec l’une des deux interventions expérimentales. Après cette période, toutes les souris traitées avec des anti-TNF sont mortes de tuberculose disséminée, avec une survie moyenne de 44 jours. Au contraire, aucune des souris traitées par abatacept n’est morte.
Alors que la concentration d’IFN-g sérique n’a pas changé dans le groupe abatacept, elle était élevée chez les souris avec anti-TNF. Cette augmentation a été attribuée à l’augmentation de l’infiltration des CD4+ et des CD8+ causée par la dispersion étendue des colonies bactériennes.
Donc, alors que les souris traitées par un anti-TNF ont montré une mortalité de 100%, l’abatacept n’a pas altéré la capacité des souris à organiser une réponse inflammatoire capable de contrôler la propagation de la tuberculose. Cependant, les données cliniques sont encore insuffisantes pour confirmer ces résultats chez l’homme.
6. Effet antirésorptif de l’abatacept sur le remodelage osseux. L’activité ostéoclastique est augmentée dans la PR, à la fois dans l’articulation, provoquant des érosions osseuses, ainsi que de manière systémique, atteignant des niveaux associés à une ostéoporose généralisée.44,45
En fait, une augmentation du ligand récepteur activateur du facteur nucléaire NF-kB (RANKL) a été mise en évidence dans la membrane synoviale.45,46 L’abatacept inhibe de manière dose-dépendante la formation d’ostéoclastes murins et l’activité ostéoclastogène évaluée in vitro. Ceci a été étudié sur des ostéoclastes murins cultivés sur des plaques de dentine, qui ont mesuré le nombre de puits de résorption après 6 jours d’ajout de différentes doses d’abatacept.47
Le médicament a significativement diminué la zone de résorption osseuse. Ces données suggèrent que l’abatacept est une molécule qui se lie directement aux cellules précurseurs des ostéoclastes, inhibant leur différenciation. Ce mécanisme pourrait expliquer l’effet anti-érosif du médicament chez les patients atteints de PR. En effet, les patients traités par abatacept ont montré une tendance à la diminution des niveaux de RANK et de son ligand RANKL dans la synovie, le tout associé à une augmentation de l’ostéoprotégérine.48 Bien que le mécanisme exact qui sous-tend cette observation ne soit pas clair, ces résultats sont bien corrélés avec l’amélioration radiologique observée chez les patients traités par abatacept.
7. Effets de l’abatacept sur d’autres cellules immunitaires. Bien que l’APC soit la cellule cible qui fixe l’abatacept et que les macrophages expriment également des récepteurs CD80/86 à leur surface, il existe peu d’études portant sur l’action du médicament sur l’activité de ces cellules. En effet, une étude in vitro récente a montré que les macrophages présentaient une expression marquée des récepteurs CD80/86 et que le traitement par l’abatacept réduisait considérablement la production de cytokines49. Ces résultats suggèrent que le mécanisme d’action du médicament pourrait être étendu à la régulation de la lignée des macrophages, cellules clés dans la pathogenèse de la maladie.
L’abatacept supprime également la migration folliculaire des cellules T spécifiques de l’antigène et, par conséquent, la collaboration entre les cellules T et les cellules B folliculaires dans le ganglion lymphatique. Cette observation a été faite in situ dans les ganglions lymphatiques de souris BALB/c50. Après avoir transfusé à ces souris des cellules T préstimulées spécifiques de l’antigène, une immunisation ultérieure des souris a montré une prolifération des cellules T et une migration vers la zone des lymphocytes B. Chez les souris traitées par abatacept, la prolifération et la migration des cellules T ont été bloquées, limitant leur présence dans la plupart des cas dans le paracortex des ganglions lymphatiques. Ainsi, un traitement prolongé par abatacept réduit la prolifération, la mobilité et la distribution des lymphocytes mémoires intraganglionnaires à auto-antigène, ce qui pourrait conduire à la diminution des auto-anticorps.
Conclusions sur le mécanisme d’action de l’abatacept
L’abatacept est une construction protéique entièrement humanisée, constituée du domaine extracellulaire de l’antigène 4 associé aux lymphocytes T cytotoxiques humains (CTL4) et d’un fragment génétiquement modifié de la région Fc des IgG1, conçu pour interférer avec la régulation de la costimulation des lymphocytes T Le médicament inhibe l’activation des cellules T en bloquant sélectivement la liaison spécifique de CD80/CD86 au récepteur CD28 et, par conséquent, en inhibant la prolifération des cellules T et les réponses immunitaires des lymphocytes B Cette action pharmacologique entraîne une diminution des niveaux de médiateurs inflammatoires chez les patients atteints de PR et une réponse clinique sûre et efficace.
Conflit d’intérêts
Le Dr Gabriel Herrero-Beaumont a reçu des subventions de recherche de Bristol-Myers-Squibb. Le Dr Santos Castañeda a reçu des subventions d’éducation et de recherche de Abbott, MSD et Pfizer.