Groupe pharmacothérapeutique : Agents antinéoplasiques, inhibiteurs de la protéine kinase, code ATC : L01XE31
Mécanisme d’action
Le nintedanib est une petite molécule inhibitrice de tyrosine kinase incluant les récepteurs du facteur de croissance dérivé des plaquettes (PDGFR) α et β, le récepteur du facteur de croissance des fibroblastes (FGFR) 1-3, et le VEGFR 1-3. En outre, le nintedanib inhibe les kinases Lck (tyrosine-protéine kinase spécifique des lymphocytes), Lyn (tyrosine-protéine kinase lyn), Src (tyrosine-protéine kinase proto-oncogène src) et CSF1R (récepteur du facteur 1 de stimulation des colonies). Le nintedanib se lie de manière compétitive à la poche de liaison de l’adénosine triphosphate (ATP) de ces kinases et bloque les cascades de signalisation intracellulaires, dont il a été démontré qu’elles sont impliquées dans la pathogenèse du remodelage tissulaire fibrotique dans les maladies pulmonaires interstitielles.
Effets pharmacodynamiques
Dans des études in vitro utilisant des cellules humaines, il a été démontré que le nintedanib inhibe les processus supposés être impliqués dans l’initiation de la pathogenèse fibrotique, la libération de médiateurs pro-fibrotiques à partir des cellules monocytaires du sang périphérique et la polarisation des macrophages en macrophages alternativement activés. Il a été démontré que le nintedanib inhibe les processus fondamentaux de la fibrose des organes, la prolifération et la migration des fibroblastes, leur transformation en phénotype myofibroblastique actif et la sécrétion de la matrice extracellulaire. Dans des études animales portant sur plusieurs modèles de FPI, de SSc/SSc-ILD, d’ILD associée à la polyarthrite rhumatoïde (PR) et de fibrose d’autres organes, le nintedanib a montré des effets anti-inflammatoires et anti-fibrotiques dans les poumons, la peau, le cœur, les reins et le foie. Le nintedanib a également exercé une activité vasculaire. Il a réduit l’apoptose des cellules endothéliales microvasculaires dermiques et atténué le remodelage vasculaire pulmonaire en réduisant la prolifération des cellules musculaires lisses vasculaires, l’épaisseur des parois des vaisseaux pulmonaires et le pourcentage de vaisseaux pulmonaires occlus.
Efficacité et sécurité cliniques
Fibrose pulmonaire idiopathique (FPI)
L’efficacité clinique du nintedanib a été étudiée chez des patients atteints de FPI dans deux études de phase III, randomisées, en double aveugle, contrôlées contre placebo, de conception identique (INPULSIS-1 (1199.32) et INPULSIS-2 (1199.34)). Les patients ayant une CVF de base < 50% prédite ou une capacité de diffusion du monoxyde de carbone (DLCO, corrigée pour l’hémoglobine) < 30% prédite au départ ont été exclus des essais. Les patients ont été randomisés dans un rapport 3:2 pour recevoir un traitement par Ofev 150 mg ou un placebo deux fois par jour pendant 52 semaines.
Le critère d’évaluation principal était le taux annuel de déclin de la capacité vitale forcée (CVF). Les principaux critères secondaires étaient le changement par rapport aux valeurs initiales du score total du questionnaire respiratoire de Saint George (SGRQ) à 52 semaines et le délai avant la première exacerbation aiguë de la FPI.
Taux annuel de déclin de la CVF
Le taux annuel de déclin de la CVF (en ml) a été significativement réduit chez les patients recevant le nintedanib par rapport aux patients recevant le placebo. L’effet du traitement était cohérent dans les deux essais. Voir le tableau 3 pour les résultats des études individuelles et groupées.
Tableau 3 : Taux annuel de déclin de la CVF (mL) dans les essais INPULSIS-1, INPULSIS-2 et leurs données regroupées – ensemble traité
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 et INPULSIS-.2 mises en commun |
||||
|
Placebo |
Ofev 150 mg deux fois par jour . |
Placebo |
Ofev 150 mg deux fois par jour |
Placebo |
Ofev 150 mg deux fois par jour |
|
|
Nombre de patients analysés |
||||||
|
Taux1 (SE) de déclin sur 52 semaines |
-239.9 (18.71) |
-114.7 (15.33) |
-207.3 (19.31) |
-113.6 (15.73) |
-223.5 (13.45) |
-113.6 (10.98) |
|
Comparaison avec le placebo |
||||||
|
Différence1 |
||||||
|
IC 95% |
(77.7, 172,8) |
(44,8, 142,7) |
(75,9, 144,0) |
|||
|
p-value |
<0.0001 |
<0,0001 |
||||
|
1 Estimé sur la base d’un modèle de régression à coefficient aléatoire. CI : intervalle de confiance |
||||||
Dans une analyse de sensibilité qui supposait que chez les patients ayant des données manquantes à la semaine 52, le déclin de la CVF après la dernière valeur observée serait le même que chez tous les patients sous placebo, la différence ajustée du taux annuel de déclin entre le nintedanib et le placebo était de 113.9 mL/an (IC à 95 % : 69,2, 158,5) dans l’étude INPULSIS-1 et de 83,3 mL/an (IC à 95 % : 37,6, 129,0) dans l’étude INPULSIS-2.
Voir la figure 1 pour l’évolution du changement par rapport à la ligne de base au fil du temps dans les deux groupes de traitement, selon l’analyse groupée des études INPULSIS-1 et INPULSIS-2.
Figure 1 : Moyenne (SEM) des changements observés de la CVF par rapport à la ligne de base (mL) au fil du temps, études INPULSIS-1 et INPULSIS-2 regroupées
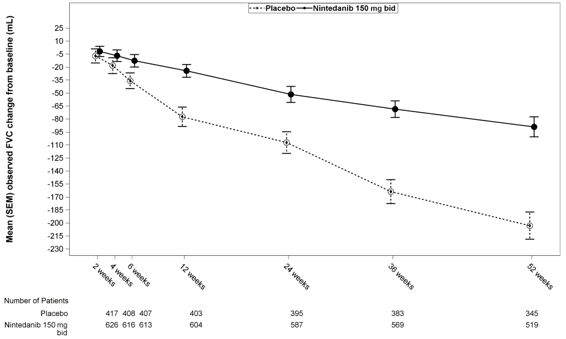
bid = deux fois par jour
Analyse des répondeurs CVF
Dans les deux essais INPULSIS, la proportion de répondeurs CVF, définie comme les patients dont le déclin absolu de la CVF % prédite ne dépassait pas 5 % (un seuil indicatif du risque croissant de mortalité dans la FPI), était significativement plus élevée dans le groupe nintedanib par rapport au placebo. Des résultats similaires ont été observés dans les analyses utilisant un seuil conservateur de 10 %. Voir le tableau 4 pour les résultats des études individuelles et groupées.
Tableau 4 : Proportion de répondeurs à la CVF à 52 semaines dans les essais INPULSIS-1, INPULSIS-2 et leurs données regroupées – ensemble traité
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 et INPULSIS-.2 mises en commun |
||||
|
Placebo |
Ofev 150 mg deux fois par jour |
Placebo |
Ofev 150 mg deux fois par jour |
Placebo |
Ofev 150 mg deux fois par jour |
|
|
Nombre de patients analysés |
||||||
|
Seuil de 5 % |
||||||
|
Nombre (%) de répondants à la CVF1 |
78 (38.2) |
163 (52.8) |
86 (39.3) |
175 (53.2) |
164 (38.8) |
338 (53.0) |
|
Comparaison avec le placebo |
||||||
|
Odds ratio |
||||||
|
IC 95% |
(1.28, 2.66) |
(1.26, 2.55) |
(1.43, 2.36) |
|||
|
p-value2 |
<0.0001 |
|||||
|
Seuil de 10% |
||||||
|
Nombre (%) de répondants à la CVF1 |
116 (56.9) |
218 (70.6) |
140 (63.9) |
229 (69.6) |
256 (60.5) |
447 (70.1) |
|
Comparaison avec le placebo |
||||||
|
Odds ratio |
||||||
|
IC 95% |
(1.32, 2.79) |
(0.89, 1.86) |
(1.21, 2.05) |
|||
|
p-value2 |
||||||
1Les patients répondeurs sont ceux qui ne présentent pas de déclin absolu supérieur à 5% ou supérieur à 10% de la CVF % prédite, selon le seuil retenu et avec une évaluation de la CVF à 52 semaines.
2Sur la base d’une régression logistique.
Temps jusqu’à la progression (déclin absolu ≥ 10% de la CVF % prédite ou décès)
Dans les deux essais INPULSIS, le risque de progression a été réduit de manière statistiquement significative pour les patients traités par nintedanib par rapport au placebo. Dans l’analyse groupée, le HR était de 0,60 indiquant une réduction de 40% du risque de progression pour les patients traités par le nintedanib par rapport au placebo.
Tableau 5 : Fréquence des patients présentant un déclin absolu ≥ 10% de la CVF % prédite ou un décès sur 52 semaines et temps jusqu’à la progression dans les essais INPULSIS-1, INPULSIS-2, et leurs données regroupées – ensemble traité
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 et INPULSIS-.2 mises en commun |
||||
|
Placebo |
Ofev 150 mg deux fois par jour |
Placebo |
Ofev 150 mg deux fois par jour |
Placebo |
Ofev 150 mg deux fois par jour |
|
|
Nombre de personnes à risque. |
||||||
|
Patients présentant des événements, N (%) |
83 (40.7) |
(24.3) |
(42.0) |
(29.8) |
(41.4) |
(27.1) |
|
Comparaison avec le placebo1 |
||||||
|
p-value2 |
<0.0001 |
|||||
|
Rapport des risques3 |
||||||
|
IC 95% |
(0,39, 0.72) |
(0,51, 0,89) |
(0,49, 0,74) |
|||
|
1 Basé sur des données collectées jusqu’à 372 jours (52 semaines + marge de 7 jours). 2 Basé sur un test Log-rank. 3 Basé sur un modèle de régression de Cox. |
||||||
Changement par rapport à la ligne de base du score total du SGRQ à la semaine 52
Dans l’analyse groupée des essais INPULSIS, les scores SGRQ de base étaient de 39,51 dans le groupe nintedanib et de 39,58 dans le groupe placebo. La variation moyenne estimée du score total du SGRQ entre le début de l’étude et la semaine 52 était plus faible dans le groupe nintedanib (3,53) que dans le groupe placebo (4,96), avec une différence entre les groupes de traitement de -1,43 (IC à 95 % : -3,09, 0,23 ; p=0,0923). Dans l’ensemble, l’effet du nintedanib sur la qualité de vie liée à la santé mesurée par le score total du SGRQ est modeste, indiquant une moindre aggravation par rapport au placebo.
Délai avant la première exacerbation aiguë de la FPI
Dans l’analyse groupée des essais INPULSIS, un risque numériquement plus faible de première exacerbation aiguë a été observé chez les patients recevant le nintedanib par rapport au placebo. Voir le tableau 6 pour les résultats des études individuelles et groupées.
Tableau 6 : Fréquence des patients présentant des exacerbations aiguës de la FPI sur 52 semaines et analyse du délai avant la première exacerbation sur la base des événements rapportés par les investigateurs dans les essais INPULSIS-1, INPULSIS-2, et leurs données regroupées – ensemble traité
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 et INPULSIS-2 mises en commun |
||||
|
Placebo |
Ofev 150 mg deux fois par jour |
Placebo |
Ofev 150 mg deux fois par jour |
Placebo |
Ofev 150 mg deux fois par jour |
|
|
Nombre à risque |
||||||
|
Patients présentant des événements, N (%) |
11 (5.4) |
19 (6.1) |
21 (9.6) |
12 (3.6) |
32 (7.6) |
31 (4.9) |
|
Comparaison avec le placebo1 |
||||||
|
p-value2 |
||||||
|
Rapport de risque3 |
||||||
|
Rapport de risque |
||||||
|
IC 95% |
(0.54, 2,42) |
(0,19, 0,77) |
(0,39, 1,05) |
|||
|
1 Basé sur des données collectées jusqu’à 372 jours (52 semaines + marge de 7 jours). 2 Basé sur un test Log-rank. 3 Basé sur un modèle de régression de Cox. |
||||||
Dans une analyse de sensibilité pré-spécifiée, la fréquence des patients avec au moins 1 exacerbation adjugée survenant dans les 52 semaines était plus faible dans le groupe nintedanib (1,9% des patients) que dans le groupe placebo (5,7% des patients). L’analyse du temps jusqu’à l’événement des exacerbations jugées en utilisant les données regroupées a donné un hazard ratio (HR) de 0,32 (IC 95 % 0,16, 0,65 ; p=0,0010).
Analyse de survie
Dans l’analyse regroupée pré-spécifiée des données de survie des essais INPULSIS, la mortalité globale sur 52 semaines était inférieure dans le groupe nintedanib (5,5 %) par rapport au groupe placebo (7,8 %). L’analyse du délai avant le décès a donné un HR de 0,70 (IC à 95 % : 0,43, 1,12 ; p=0,1399). Les résultats de tous les critères de survie (tels que la mortalité pendant le traitement et la mortalité respiratoire) ont montré une différence numérique constante en faveur du nintedanib.
Tableau 7 : Mortalité toutes causes confondues sur 52 semaines dans les essais INPULSIS-1, INPULSIS-2, et leurs données regroupées – ensemble traité
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 et INPULSIS-2 mises en commun |
||||
|
Placebo |
Ofev 150 mg deux fois par jour |
Placebo |
Ofev 150 mg deux fois par jour |
Placebo |
Ofev 150 mg deux fois par jour |
|
|
Nombre à risque |
||||||
|
Patients présentant des événements, N (%) |
13 (6.4) |
13 (4.2) |
20 (9.1) |
22 (6.7) |
33 (7.8) |
35 (5.5) |
|
Comparaison avec le placebo1 |
||||||
|
p-value2 |
||||||
|
Rapport de risque3 |
||||||
|
Rapport de risque |
||||||
|
IC 95% |
(0.29, 1,36) |
(0,40, 1,35) |
(0,43, 1,12) |
|||
|
1 Basé sur des données collectées jusqu’à 372 jours (52 semaines + marge de 7 jours). 2 Basé sur un test Log-rank. 3 Sur la base d’un modèle de régression de Cox. |
||||||
Traitement à long terme avec Ofev chez les patients atteints de FPI (INPULSIS-ON)
Un essai d’extension ouvert d’Ofev a inclus 734 patients atteints de FPI. Les patients qui ont terminé la période de traitement de 52 semaines dans un essai INPULSIS ont reçu un traitement Ofev en ouvert dans l’essai d’extension INPULSIS-ON. La durée médiane d’exposition des patients traités par Ofev dans les essais INPULSIS et INPULSIS-ON était de 44,7 mois (intervalle de 11,9 à 68,3). Les paramètres d’efficacité exploratoires comprenaient le taux annuel de déclin de la CVF sur 192 semaines, qui était de -135,1 (5,8) ml/an chez tous les patients traités et qui correspondait au taux annuel de déclin de la CVF chez les patients traités par Ofev dans les essais de phase III de l’INPULSIS (-113,6 ml par an). Le profil d’effets indésirables d’Ofev dans l’étude INPULSIS-ON était cohérent avec celui des essais de phase III INPULSIS.
Patients atteints de FPI avec une altération avancée de la fonction pulmonaire (INSTAGE)
INSTAGE était un essai clinique multicentrique, multinational, prospectif, randomisé, en double aveugle et en groupes parallèles chez des patients atteints de FPI avec une altération avancée de la fonction pulmonaire (DLCO ≤ 35% prédite) pendant 24 semaines. 136 patients ont été traités par Ofev en monothérapie. Le résultat du critère d’évaluation primaire a montré une réduction du score total du questionnaire respiratoire de St Georges (SGRQ) de -0,77 unité à la semaine W12, sur la base du changement moyen ajusté par rapport à la ligne de base. Une comparaison post hoc a démontré que le déclin de la CVF chez ces patients était cohérent avec le déclin de la CVF chez les patients présentant une maladie moins avancée et traités par Ofev dans les essais de phase III de l’INPULSIS.
Le profil de sécurité et de tolérance d’Ofev chez les patients atteints de FPI présentant une altération avancée de la fonction pulmonaire était cohérent avec celui observé dans les essais de phase III de l’INPULSIS.
Données supplémentaires de l’essai de phase IV INJOURNEY avec Ofev 150 mg deux fois par jour et ajout de pirfénidone
Le traitement concomitant avec le nintedanib et la pirfénidone a été étudié dans un essai exploratoire ouvert, Le traitement concomitant par le nintedanib et la pirfénidone a été étudié dans le cadre d’un essai exploratoire ouvert et randomisé portant sur l’association de 150 mg de nintedanib deux fois par jour et d’un complément de pirfénidone (titré à 801 mg trois fois par jour), comparé au nintedanib 150 mg deux fois par jour seul, chez 105 patients randomisés pendant 12 semaines. Le critère d’évaluation principal était le pourcentage de patients présentant des effets indésirables gastro-intestinaux entre le début de l’étude et la semaine 12. Les effets indésirables gastro-intestinaux étaient fréquents et conformes au profil de sécurité établi de chaque composant. La diarrhée, les nausées et les vomissements ont été les événements indésirables les plus fréquents rapportés chez les patients, traités par la pirfénidone ajoutée au nintedanib par rapport au nintedanib seul, respectivement.
Les changements absolus moyens (SE) par rapport à la ligne de base de la CVF à la semaine 12 étaient de -13,3 (17,4) ml chez les patients traités par le nintedanib avec la pirfénidone ajoutée (n=48) par rapport à -40,9 (31,4) ml chez les patients traités par le nintedanib seul (n=44).
Autres maladies pulmonaires interstitielles (MPI) chroniques fibrosantes à phénotype progressif
L’efficacité clinique d’Ofev a été étudiée chez des patients atteints d’autres MPI chroniques fibrosantes à phénotype progressif dans une étude de phase III en double aveugle, randomisée, contrôlée par placebo (INBUILD). Les patients atteints de FPI ont été exclus. Les patients avec un diagnostic clinique d’une ILD chronique fibrosante ont été sélectionnés s’ils présentaient une fibrose pertinente (plus de 10% de caractéristiques fibrotiques) sur HRCT et présentaient des signes cliniques de progression (définis comme un déclin de la CVF ≥10%, un déclin de la CVF ≥ 5% et <10% avec une aggravation des symptômes ou de l’imagerie, ou une aggravation des symptômes et une aggravation de l’imagerie, le tout dans les 24 mois précédant le dépistage). Les patients devaient avoir une CVF supérieure ou égale à 45% de la valeur prédite et une DLCO de 30% à moins de 80% de la valeur prédite. Les patients devaient avoir progressé malgré une prise en charge jugée appropriée en pratique clinique pour l’ILD concernée.
Un total de 663 patients ont été randomisés dans un rapport 1:1 pour recevoir soit Ofev 150 mg bid soit un placebo correspondant pendant au moins 52 semaines. L’exposition médiane à Ofev sur l’ensemble de l’essai était de 17,4 mois et l’exposition moyenne à Ofev sur l’ensemble de l’essai était de 15,6 mois. La randomisation a été stratifiée sur la base du modèle fibrotique HRCT évalué par des lecteurs centraux. 412 patients présentant une HRCT avec un profil fibrotique de type pneumonie interstitielle habituelle (UIP) et 251 patients présentant d’autres profils fibrotiques HRCT ont été randomisés. Deux populations coprimaires ont été définies pour les analyses de cet essai : tous les patients (la population globale) et les patients présentant une HRCT avec un profil fibrotique de type UIP. Les patients présentant d’autres profils fibrotiques HRCT représentaient la population « complémentaire ».
Le critère d’évaluation principal était le taux annuel de déclin de la capacité vitale forcée (CVF) (en ml) sur 52 semaines. Les principaux critères d’évaluation secondaires étaient le changement absolu par rapport à la ligne de base du score total du King’s Brief Interstitial Lung Disease Questionnaire (K-BILD) à la semaine 52, le temps jusqu’à la première exacerbation aiguë de l’ILD ou le décès sur 52 semaines, et le temps jusqu’au décès sur 52 semaines.
Les patients avaient un âge moyen (écart-type) de 65,8 (9,8, 27-87) ans et un pourcentage moyen de CVF prédite de 69,0% (15,6, 42-137). Les diagnostics cliniques sous-jacents des ILD dans les groupes représentés dans l’essai étaient la pneumopathie d’hypersensibilité (26,1 %), les ILD auto-immunes (25,6 %), la pneumonie interstitielle idiopathique non spécifique (18,9 %), la pneumonie interstitielle idiopathique inclassable (17,2 %) et d’autres ILD (12,2 %).
L’essai INBUILD n’était pas conçu ou alimenté pour fournir des preuves d’un bénéfice du nintedanib dans des sous-groupes de diagnostic spécifiques. Des effets cohérents ont été démontrés dans les sous-groupes basés sur les diagnostics d’ILD. L’expérience avec le nintedanib dans les ILD fibrosantes progressives très rares est limitée.
Taux annuel de déclin de la CVF
Le taux annuel de déclin de la CVF (en ml) sur 52 semaines a été significativement réduit de 107,0 ml chez les patients recevant Ofev par rapport aux patients recevant le placebo (tableau 8) correspondant à un effet relatif du traitement de 57,0%.
Tableau 8 : Taux annuel de déclin de la CVF (mL) sur 52 semaines
|
Placebo |
Ofev 150 mg deux fois par jour |
|
|
Nombre de patients analysés |
||
|
Taux1 (SE) de déclin sur 52 semaines |
-.187.8 (14.8) |
-80.8 (15.1) |
|
Comparaison avec le placebo |
||
|
Différence1 |
||
|
IC95% |
(65.4, 148,5) |
|
|
valeurp |
< 0.0001 |
|
1Sur la base d’une régression à coefficient aléatoire avec des effets catégoriels fixes du traitement, du modèle HRCT, des effets continus fixes du temps, de la CVF de base, et incluant des interactions traitement par temps et base par temps
Des résultats similaires ont été observés dans la population coprimaire de patients avec HRCT avec un modèle fibrotique de type UIP. L’effet du traitement était cohérent dans la population complémentaire de patients présentant d’autres profils fibrotiques HRCT (valeur de p d’interaction 0,2268) (Figure 2).
Figure 2 Graphique en forêt du taux annuel de déclin de la CVF (mL) sur 52 semaines dans les populations de patients
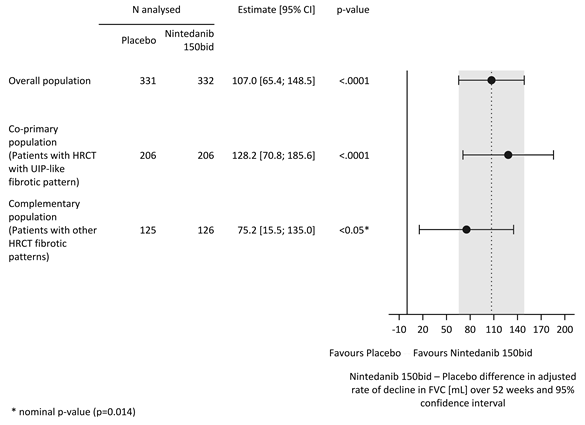
bid = deux fois par jour
Les résultats de l’effet d’Ofev sur la réduction du taux annuel de déclin de la CVF ont été confirmés par toutes les analyses de sensibilité pré-spécifiées et des résultats cohérents ont été observés dans les sous-groupes d’efficacité pré-spécifiés : le sexe, le groupe d’âge, la race, la CVF prédite de base en %, et le diagnostic clinique sous-jacent original de l’ILD dans les groupes.
La figure 3 montre l’évolution du changement de la CVF par rapport à la ligne de base au fil du temps dans les groupes de traitement.
Figure 3 Moyenne (SEM) du changement observé de la CVF par rapport à la ligne de base (mL) sur 52 semaines
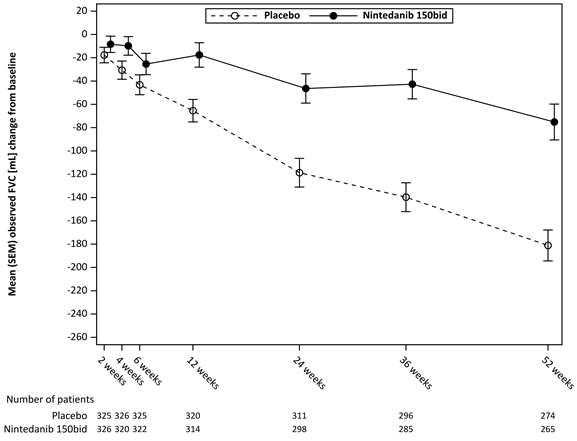
bid = deux fois par jour
En outre, des effets favorables d’Ofev ont été observés sur le changement absolu moyen ajusté par rapport à la ligne de base de la CVF % prédite à la semaine 52. Le changement absolu moyen ajusté de la base à la semaine 52 de la CVF % prédite était plus faible dans le groupe nintedanib (-2,62%) que dans le groupe placebo (-5,86%). La différence moyenne ajustée entre les groupes de traitement était de 3,24 (IC 95 % : 2,09, 4,40, p<0,0001 nominal).
Analyse des répondeurs CVF
La proportion de répondeurs CVF, définis comme des patients présentant une baisse relative de la CVF % prédite ne dépassant pas 5 %, était plus élevée dans le groupe Ofev par rapport au groupe placebo. Des résultats similaires ont été observés dans les analyses utilisant un seuil de 10% (tableau 9).
Tableau 9 : Proportion de répondeurs à la CVF à 52 semaines dans INBUILD
|
Placebo |
Ofev 150 mg deux fois par jour |
|
|
Nombre de patients analysés |
||
|
5% seuil |
||
|
Nombre (%) de répondeurs FVC1 |
104 (31.4) |
158 (47.6) |
|
Comparaison avec le placebo |
||
|
Odds ratio² |
||
|
IC 95% |
(1.46, 2,76) |
|
|
Valeur nominale de p |
< 0.0001 |
|
|
Seuil de 10% |
||
|
Nombre (%) de répondants à la CVF1 |
169 (51.1) |
197 (59.3) |
|
Comparaison avec le placebo |
||
|
Odds ratio² |
||
|
IC 95% |
(1,04, 1.94) |
|
|
Valeur p nominale |
||
1Les patients répondeurs sont ceux qui n’ont pas de déclin relatif supérieur à 5% ou supérieur à 10% de la CVF % prédite, selon le seuil, et qui ont une évaluation de la CVF à 52 semaines (les patients avec des données manquantes à la semaine 52 ont été considérés comme non répondeurs).
2Sur la base d’un modèle de régression logistique avec covariable continue CVF % prédite de base et covariable binaire schéma HRCT
Temps avant la première exacerbation aiguë de l’ILD ou le décès
Sur l’ensemble de l’essai, la proportion de patients avec au moins un événement de première exacerbation aiguë de l’ILD ou de décès était de 13,9% dans le groupe Ofev et de 19,6% dans le groupe placebo. Le HR était de 0,67 (IC 95 % : 0,46, 0,98 ; p nominal=0,0387), indiquant une réduction de 33 % du risque de première exacerbation aiguë de la PI ou de décès chez les patients recevant Ofev par rapport au placebo (figure 4).
Figure 4 Tracé de Kaplan-Meier du délai avant la première exacerbation aiguë de l’ILD ou le décès sur l’ensemble de l’essai
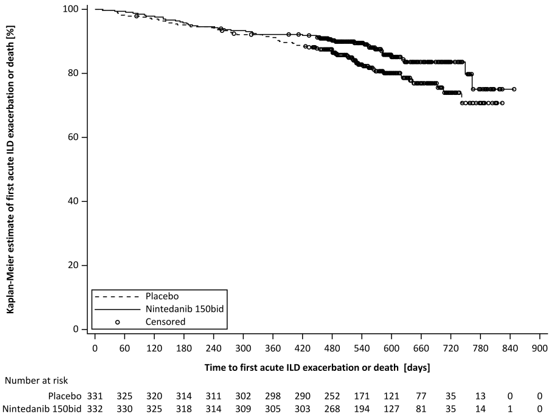
bid = deux fois par jour
Analyse de survie
Le risque de décès était inférieur dans le groupe Ofev par rapport au groupe placebo. Le HR était de 0,78 (IC 95 % : 0,50, 1,21 ; p nominal=0,2594), indiquant une réduction de 22 % du risque de décès chez les patients recevant Ofev par rapport au placebo.
Temps jusqu’à la progression (déclin absolu ≥ 10 % de la CVF % prédite) ou le décès
Dans l’essai INBUILD, le risque de progression (déclin absolu ≥ 10 % de la CVF % prédite) ou de décès était réduit pour les patients traités par Ofev. La proportion de patients présentant un événement était de 35,2% dans le groupe Ofev et de 48,3% dans le groupe placebo. Le HR était de 0,66 (IC 95 % : 0,53, 0,83 ; p=0,0003), indiquant une réduction de 34 % du risque de progression (déclin absolu ≥ 10 % de la CVF % prédite) ou de décès chez les patients recevant Ofev par rapport au placebo.
Qualité de vie
Le changement moyen ajusté par rapport à la ligne de base du score total K-BILD à la semaine 52 était de -0,79 unité dans le groupe placebo et de 0,55 dans le groupe Ofev. La différence entre les groupes de traitement était de 1,34 (IC 95 % : -0,31, 2,98 ; p nominal=0,1115).
Le changement absolu moyen ajusté par rapport au début de l’étude du score du domaine dyspnée des symptômes Living with Pulmonary Fibrosis (L-PF) à la semaine 52 était de 4,28 dans le groupe Ofev contre 7,81 dans le groupe placebo. La différence moyenne ajustée entre les groupes en faveur d’Ofev était de -3,53 (IC 95 % : -6,14, -0,92 ; p nominal=0,0081). Le changement absolu moyen ajusté par rapport à la ligne de base du score du domaine toux de L-PF Symptômes à la semaine 52 était de -1,84 dans le groupe Ofev contre 4,25 dans le groupe placebo. La différence moyenne ajustée entre les groupes en faveur d’Ofev était de -6,09 (IC à 95 % : -9,65, -2,53 ; p nominal=0,0008).
Pneumopathie interstitielle associée à la sclérose systémique (SSc-ILD)
L’efficacité clinique d’Ofev a été étudiée chez des patients atteints de SSc-ILD dans le cadre d’une étude de phase III en double aveugle, randomisée et contrôlée par placebo (SENSCIS). Les patients ont été diagnostiqués comme atteints de SSc-ILD sur la base des critères de classification 2013 de l’American College of Rheumatology / Ligue européenne contre le rhumatisme pour la SSc et d’une tomographie thoracique à haute résolution (HRCT) réalisée dans les 12 mois précédents. Au total, 580 patients ont été randomisés dans un groupe de 1 :1 pour recevoir soit Ofev 150 mg bid, soit un placebo correspondant pendant au moins 52 semaines. 576 patients ont été traités. La randomisation a été stratifiée en fonction du statut des anticorps anti-topoisomérase (ATA). Les patients sont restés sous traitement en aveugle jusqu’à 100 semaines (exposition médiane à l’Ofev : 15,4 mois ; exposition moyenne à l’Ofev : 14,5 mois).
Le critère d’évaluation principal était le taux annuel de déclin de la CVF sur 52 semaines. Les principaux critères d’évaluation secondaires étaient le changement absolu par rapport à la ligne de base du score cutané modifié de Rodnan (mRSS) à la semaine 52 et le changement absolu par rapport à la ligne de base du score total du questionnaire respiratoire de Saint George (SGRQ) à la semaine 52.
Dans la population globale, 75,2% des patients étaient des femmes. L’âge moyen (écart-type ) était de 54,0 (12,2, 20-79) ans. Dans l’ensemble, 51,9% des patients avaient une sclérose systémique (SSc) cutanée diffuse et 48,1% une SSc cutanée limitée. Le délai moyen (écart-type) depuis la première apparition d’un symptôme non-Raynaud était de 3,49 (1,7) ans. 49,0 % des patients suivaient un traitement stable par mycophénolate au début de l’étude. Le profil de sécurité chez les patients avec ou sans mycophénolate au départ était comparable.
Taux annuel de déclin de la CVF
Le taux annuel de déclin de la CVF (ml) sur 52 semaines a été significativement réduit de 41,0 ml chez les patients recevant Ofev par rapport aux patients recevant le placebo (tableau 10) correspondant à un effet relatif du traitement de 43,8%.
Tableau 10 : Taux annuel de déclin de la CVF (mL) sur 52 semaines
|
Placebo |
Ofev 150 mg deux fois par jour |
|
|
Nombre de patients analysés |
||
|
Taux1 (SE) de déclin sur 52 semaines |
-.93.3 (13.5) |
-52.4 (13.8) |
|
Comparaison avec le placebo |
||
|
Différence1 |
||
|
IC à 95% |
(2.9, 79.0) |
|
|
p-value |
<0.05 |
1Sur la base d’une régression à coefficient aléatoire avec des effets catégoriels fixes du traitement, du statut ATA, du sexe, des effets continus fixes du temps, de la CVF de base, de l’âge, de la taille, et incluant des interactions traitement-temps et base-temps. Un effet aléatoire a été inclus pour l’interception et le temps spécifiques au patient. Les erreurs intra-patient ont été modélisées par une matrice de variance-covariance non structurée. La variabilité interindividuelle a été modélisée par une matrice de variance-covariance à composantes.
L’effet d’Ofev sur la réduction du taux annuel de déclin de la CVF était similaire dans les analyses de sensibilité pré-spécifiées et aucune hétérogénéité n’a été détectée dans les sous-groupes pré-spécifiés (par exemple selon l’âge, le sexe et l’utilisation du mycophénolate).
En outre, des effets similaires ont été observés sur d’autres paramètres de la fonction pulmonaire, par exemple le changement absolu par rapport à la ligne de base de la CVF en mL à la semaine 52 (Figure 5 et Tableau 11) et le taux de déclin de la CVF en % prédit sur 52 semaines (Tableau 12) fournissant une justification supplémentaire des effets d’Ofev sur le ralentissement de la progression de la SSc-ILD. En outre, moins de patients dans le groupe Ofev ont présenté un déclin absolu de la CVF > 5% de la valeur prédite (20,6% dans le groupe Ofev contre 28,5% dans le groupe placebo, OR=0,65, p=0,0287). Le déclin relatif de la CVF en mL > 10% était comparable entre les deux groupes (16,7% dans le groupe Ofev vs 18,1% dans le groupe placebo, OR=0,91, p=0,6842). Dans ces analyses, les valeurs de CVF manquantes à la semaine 52 ont été imputées avec la pire valeur du patient sous traitement.
Une analyse exploratoire des données jusqu’à 100 semaines (durée maximale du traitement dans SENSCIS) a suggéré que l’effet sur traitement d’Ofev sur le ralentissement de la progression de la SSc-ILD persistait au-delà de 52 semaines.
Figure 5 : Moyenne (SEM) du changement observé de la CVF par rapport à la ligne de base (mL) sur 52 semaines
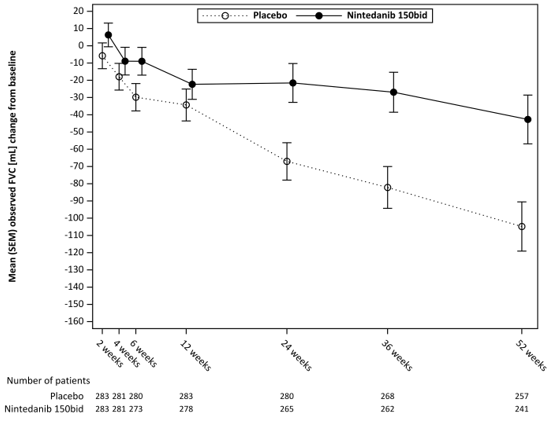
bid = deux fois par jour
Tableau 11 : Variation absolue de la CVF (mL) par rapport aux valeurs initiales à la semaine 52
|
Placebo |
Ofev 150 mg deux fois par jour . |
|
|
Nombre de patients analysés |
||
|
Moyenne (ET) au départ |
2541.0 (815,5) |
2458,5 (735,9) |
|
Moyenne1 (ET) changement par rapport à la ligne de base à la semaine 52 |
-101,0 (13,6) |
-54,6 (13.9) |
|
Comparaison avec le placebo |
||
|
Moyenne1 |
||
|
IC 95% |
(8.1, 84,7) |
|
|
p-value |
<0.05 |
1Selon le modèle mixte à mesures répétées (MMRM), avec des effets catégoriels fixes du statut ATA, de la visite, de l’interaction traitement-visite, de l’interaction base-visite : âge, sexe et taille. La visite était la mesure répétée. Les erreurs intra-patient ont été modélisées par une structure de variance-covariance non structurée. La moyenne ajustée était basée sur tous les patients analysés dans le modèle (pas seulement les patients avec une ligne de base et une mesure à la semaine 52).
Tableau 12 : Taux annuel de déclin de la CVF (% prédit) sur 52 semaines
|
Placebo |
Ofev 150 mg deux fois par jour . |
|
|
Nombre de patients analysés |
||
|
Rate1 (SE) de déclin sur 52 semaines |
-.2.6 (0.4) |
-1.4 (0.4) |
|
Comparaison avec le placebo |
||
|
Différence1 |
||
|
IC à 95% |
(0.09, 2.21) |
|
|
p-value |
<0,05 |
1Sur la base d’une régression à coefficient aléatoire avec des effets catégoriels fixes du traitement, du statut ATA, des effets continus fixes du temps, de la CVF de base, et incluant des interactions traitement-temps et base-temps. Un effet aléatoire a été inclus pour l’interception et le temps spécifiques au patient. Les erreurs intra-patient ont été modélisées par une matrice de variance-covariance non structurée. La variabilité inter-individuelle a été modélisée par une matrice de variance-covariance à composantes
Changement par rapport à la ligne de base du score cutané modifié de Rodnan (mRSS) à la semaine 52
Le changement absolu moyen ajusté par rapport à la ligne de base du mRSS à la semaine 52 était comparable entre le groupe Ofev (-2,17 (IC 95 % -2,69, -1,65)) et le groupe placebo (-1,96 (IC 95 % -2,48, -1,45)). La différence moyenne ajustée entre les groupes de traitement était de -0,21 (IC 95 % -0,94, 0,53 ; p = 0,5785).
Changement par rapport à la ligne de base dans le St. George’s Respiratory Questionnaire (SGRQ) à la semaine 52
Le changement absolu moyen ajusté par rapport au début de l’étude du score total du SGRQ à la semaine 52 était comparable entre le groupe Ofev (0,81 (IC à 95 % -0,92, 2,55)) et le groupe placebo (-0,88 (IC à 95 % -2,58, 0,82)). La différence moyenne ajustée entre les groupes de traitement était de 1,69 (IC 95% -0,73, 4,12 ; p = 0,1711).
Analyse de survie
La mortalité sur l’ensemble de l’essai était comparable entre le groupe Ofev (N = 10 ; 3,5%) et le groupe placebo (N = 9 ; 3,1%). L’analyse du temps jusqu’au décès sur l’ensemble de l’essai a donné un HR de 1,16 (IC 95 % 0,47, 2,84 ; p = 0,7535).
Intervalle QT
Dans une étude dédiée chez des patients atteints de cancer des cellules rénales, des mesures de QT/QTc ont été enregistrées et ont montré qu’une dose orale unique de 200 mg de nintedanib ainsi que des doses orales multiples de 200 mg de nintedanib administrées deux fois par jour pendant 15 jours n’ont pas prolongé l’intervalle QTcF.
Population pédiatrique
L’Agence européenne des médicaments a renoncé à l’obligation de soumettre les résultats des études avec Ofev dans tous les sous-ensembles de la population pédiatrique dans la FPI (voir section 4.2 pour les informations sur l’utilisation pédiatrique).