SONAL DESAI, ARCHITA PATEL ÉS S. Y. GABHE*
C. U. Shah Gyógyszerészeti Főiskola, S. N. D. T. Női Egyetem, Sir Vithaldas Vidya Vihar, Juhu Tara Road, Santacruz (W), Mumbai – 400 049, India
Levelező szerző: S. Y. GABHE E-mail: S. Y:
| Az elfogadás dátuma | 16-Jan-2011 | |
| A felülvizsgálat dátuma | 26- | .Oct-2010 |
| Date of Received | 9-Apr-2009 |
DOI: 10.4103/0250-474X.89762
Abstract
Egyszerű izokratikus fordított fázisú nagy teljesítményű folyadékkromatográfiát alkalmaztak a 8-klórtetofillin mintában jelen lévő három szennyeződés elválasztására. A szennyeződések jellemzésére LC-MS-t használtunk. A tömegspektrális adatok alapján e szennyeződések szerkezetét 3,7-dihidro-1,3-dimetil-1H-purin-2,6-dionként (szennyeződés I), 3,7-dihidro-1,3,7-trimetil-1H-purin-2,6-dionként (szennyeződés II) és a 8-klór-1,3-dimetil-2,6(3H,1H)-purindion izomerjeként (szennyeződés III) jellemezték.
Kulcsszavak
8-klórtheofillin, szennyeződés, LC-MS, fordított fázisú HPLC, teofillin
Bevezetés
A tömeges gyógyszeripar minden gyógyszeripar alapját képezi, mivel ez a meghatározott minőségű gyógyszerhatóanyagok (API-k) forrása. Az ömlesztett gyógyszeripar számára a legnagyobb kihívást az jelenti, hogy gazdaságosan állítsa elő a kívánt minőségű és tisztaságú végleges gyógyszert. A hatóanyagok tisztasága számos tényezőtől függ, például a nyersanyagoktól, a gyártási módszerektől és a kristályosítási vagy tisztítási eljárás típusától. Teljesen tiszta anyagokat azonban szinte lehetetlen előállítani, mivel a gyártás, a tisztítás vagy a tárolás során szennyeződések kerülnek bele. A szennyeződés a hatóanyag bármely olyan összetevője (a víz kivételével), amely nem a hatóanyagként meghatározott kémiai egység. A kémiai szintézissel előállított gyógyszerhatóanyagok esetében az ICH a szennyeződéseket három kategóriába sorolja: szerves szennyeződések, szervetlen szennyeződések és oldószer-maradékok. Az anyagban lévő szennyeződések lehetnek toxikusak, megváltoztathatják az anyag fizikai vagy kémiai tulajdonságait, ezáltal gyógyászatilag használhatatlanná téve azt. A szennyeződések csökkenthetik a termék eltarthatósági idejét, és nehézségeket okozhatnak a formulázásban. Ezért nemcsak a szennyeződések ellenőrzése, hanem a szennyeződések minősítése is kritikus kérdés az ömlesztett gyógyszeripar számára.
Az antihisztaminok, mint például a dimenhidrinát és a prometazin teokláth széles körben használt gyógyszerek az utazási betegség kezelésében. A 8-klórtheofillin, kémiailag 8-klór-1,3-dimetil-2,6(1H, 3H)- purindion, egy köztes termék, amelyet e gyógyszerek sóformájának előállításához használnak. Lényeges a dimenhidrinát és a prometazin-teokláth tisztaságának és biztonságosságának biztosítása érdekében. Ennek érdekében a 8-klór-otheofillint a legnagyobb tisztasággal és ismert szennyeződésprofillal kell előállítani.
Az irodalmi áttekintésből kiderül, hogy Wadke és munkatársai a 9-metilizoalloxazin és a 3,9-dimetilizoalloxazin kölcsönhatásait vizsgálták 8-klór-otheofillinnel. A 8-klórtotheofillint belső standardként használták az urát HPLC-módszerrel történő meghatározásához. Potentiometrikusan is meghatározták. A klórfenoxamin-hidroklorid, a 8-klórtetofillin és a koffein egyidejű meghatározását többkomponensű gyógyszerformában vékonyréteg-kromatográfiás-denzitometriás módszerrel végeztük. Gil és munkatársai ciklikus voltammetriával és differenciális impulzus-polarográfiával vizsgálták a 8-klórtotheofillin elektroanalitikai viselkedését, és differenciális impulzus-polarográfiával határozták meg gyógyszerészeti készítményekben lévő tartalmát. Stabilitást jelző RP-HPLC-módszert fejlesztettek ki és validáltak a 8-klórtotheofillinre, valamint a difenhidraminra és a koffeinre. Arány-spektroszkópiás nullpontos első derivált spektrofotometriás és kemometriai módszereket is kidolgoztunk a koffein, a 8-klórtotheofillin és a klórfenoxamin-hidroklorid egyidejű meghatározására háromkomponensű keverékekben. Eddig nem számoltak be kromatográfiás és spektroszkópiai módszerről a 8-klórtotheofillinben jelen lévő szennyeződések elválasztására és jellemzésére. Ezért a jelen munka célja a 8-klórtotheofillinben jelen lévő szennyeződések elkülönítése és jellemzése volt modern analitikai technikák alkalmazásával.
A 8-klórtotheofillin a Kores (India) Ltd., Thane ajándék mintája volt. Az összes többi vegyszert és reagenseket az S. D. Fine Chemicals Ltd-től (Mumbai, India) szereztük be. A TLC és preparatív TLC vizsgálatokhoz használt oldószerek analitikai minőségűek, a HPLC vizsgálatokhoz használt oldószerek pedig HPLC minőségűek voltak. A puffer előállításához AR minőségű nátrium-acetát-trihidrátot használtunk.
Előzetesen TLC-vizsgálatokat végeztünk a mintában jelen lévő szennyeződések számának megismerése érdekében. Állófázisként szilikagél 60GF254 (Merck) előzetesen bevont TLC lemezeket használtunk. A mintát minimális mennyiségű etil-acetátban feloldottuk, és ezt az oldatot használtuk a TLC-lemezek foltozásához. Különböző mobil fázisokat próbáltunk ki. Az etil-acetát:toluol:jégecetsav (10:0,3:0,5 v/v/v) jobb elválasztást mutatott, mint a többi mobilfázis. A 8-klór-otheofillin mintából négy komponenst választottunk el TLC-vel, az Rf értékek 0,029, 0,132, 0,198 és 0,852 voltak. A 8-klórtotheofillin Rf értéke 0,852 volt.
Amikor a TLC-hez szükséges mobilfázist kifejlesztették, kísérletet tettek a keverék preparatív TLC-vel történő elválasztására. A mintát minimális mennyiségű etil-acetátban oldottuk fel, és sávos formában foltoztuk. Mindhárom szennyeződést preparatív TLC-vel választottuk el a 8-klórtetofillintől, a TLC-vizsgálatok során alkalmazott kromatográfiás feltételek alkalmazásával. A különböző sávokat külön gyűjtöttük össze és etil-acetát segítségével extraháltuk. Mivel az egyes izolált I., II. és III. szennyeződések mennyisége nagyon kicsi volt, úgy döntöttünk, hogy ezeket a szennyeződéseket együttesen izoláljuk. Az I., II. és III. szennyeződést együttesen I. frakciónak neveztük el. Az I. frakciót preparatív TLC-vel izoláltuk a 8-klórtetofillinből. Mivel az egyes szennyeződéseket nem külön-külön izolálták, a különböző azonosítási technikákat, mint például IR, NMR, nem lehetett elvégezni. Úgy döntöttünk, hogy a további vizsgálatokat LC-MS segítségével végezzük el, amely lehetővé teszi az egyidejű elválasztást és jellemzést. Az LC-MS-elemzést megelőzően az I. frakcióhoz HPLC-profilt alakítottak ki. A HPLC-vizsgálatokhoz egy CCPM dugattyús szivattyúval, PX8010 szivattyúvezérlővel és UV-detektorral felszerelt Tosoh nagy teljesítményű folyadékkromatográfot használtak. Az injektálószelepes egységhez 20 µl kapacitású hurkot illesztettünk. Az I. frakciót acetonitrilben oldottuk, és fordított fázisú HPLC-analízisnek vetettük alá acetonitril: nátrium-acetát-trihidrát (pH 3,57; 0,01 M) (5:95 v/v) mobilfázissal. A kiválasztott oszlop Phenomenex ODS (250×4,6 mm I.D.; részecskeméret 5 µm) volt. Az áramlási sebesség 1,5 ml/perc volt, a detektálás 280 nm-es hullámhosszon történt. A mozgófázist felhasználás előtt vákuumban G5 szinterezett üvegszűrőn keresztül szűrtük, és a légbuborékok eltávolítása érdekében szonikáztuk. Az I. frakció HPLC-elemzése három csúcsot is kimutatott, amelyek átlagos retenciós ideje 8,324 perc, 14,275 perc és 20,933 perc volt (1. ábra). A 2,086 perc átlagos retenciós idővel rendelkező csúcs volt az oldószercsúcs.
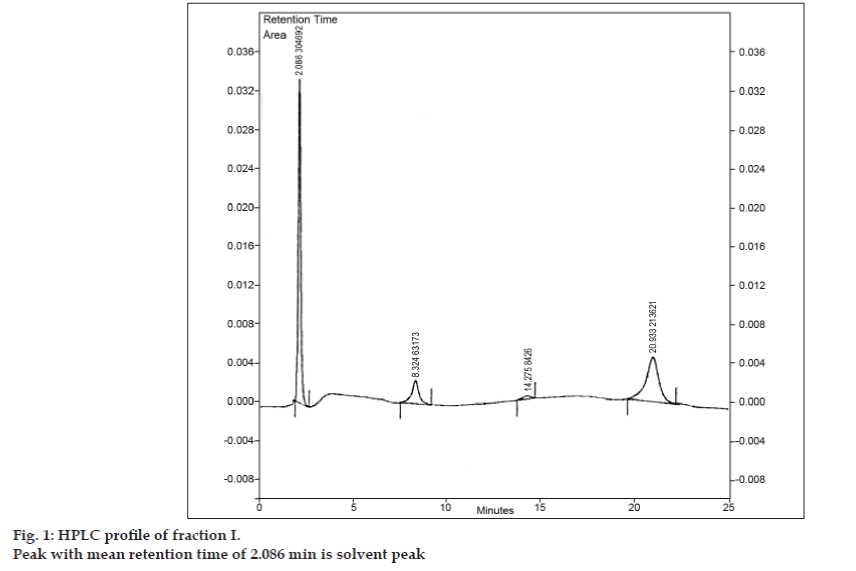
1. ábra: Az I. frakció HPLC-profilja.
Az I. frakciót ezután LC-MS analízisnek vetettük alá a szennyeződések jellemzésére. Az LC-MS vizsgálatokat egy olyan rendszeren végeztük, amelynek LC része 1100-as sorozatú HPLC (Agilent Technologies, USA) volt, amely vákuummegszűrőből (G1322A), kvaterner szivattyúból (G1311A), automatikus mintavevőből (G1313A) és UV/látható detektorból (G1314A) állt, MS része pedig háromszoros kvadrupolos tömegspektrométer Quattro II (Micromass UK Ltd.) volt, UK).
Az LC-MS vizsgálatokban a folyadékkromatográfiás elválasztást Phenomenex C18 oszlopon (250×4,6 mm, 5 µm) végeztük szobahőmérsékleten, acetonitril: nátrium-acetát puffer (5:95, v/v) mobilfázisban, 1,5 ml/perc áramlási sebességgel. A tömegspektrométert pozitív elektronporlasztásos ionizációs (ESI) üzemmódban működtettük, a tömeg/töltés (m/z) arány 120-500 m/z tartományban volt. Porlasztógázként nitrogént használtunk. Az adatokat a Masslynx szoftverrel vettük fel és dolgoztuk fel. A szennyeződések tömegspektrális adatait kaptuk (2., 3. és 4. ábra). A három csúcs fragmentációs útját a metilcsoport és/vagy a karbonilcsoport elvesztése jellemzi. A 180,9, 195,7 és 214,9 m/z értékű csúcsok +, + és + csúcsoknak felelnek meg. A kapott MS-adatok szerint a 8,324 percnél, 14,275 percnél és 20,933 percnél eluálódó szennyeződések a teofillin (mol. tömeg 180), a koffein (mol. tömeg 194) és a 8-klórteofillin izomerje (mol. tömeg 214,5) voltak (1. táblázat). Így három szennyeződést különítettünk el, és szerkezetüket a tömegspektrális adatok alapján tisztáztuk (1. ábra).
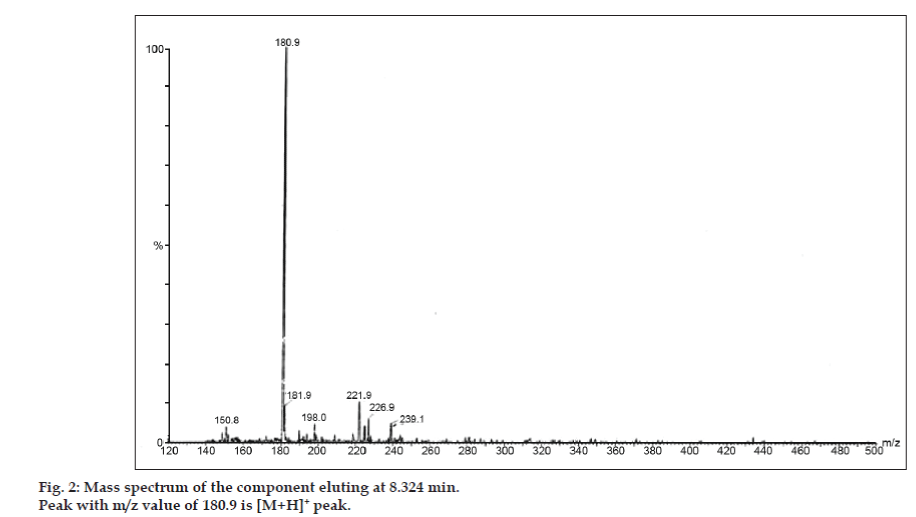
2. ábra: A 8,324 percnél eluálódó komponens tömegspektruma.
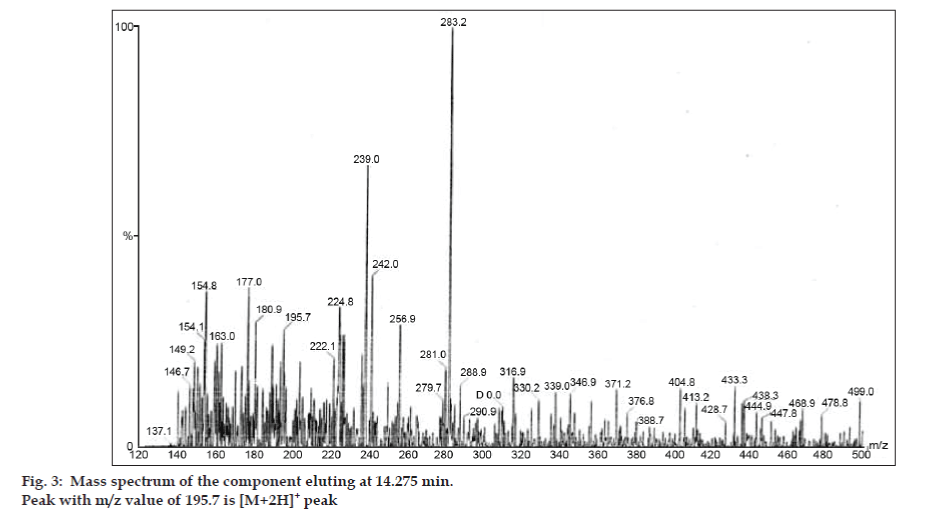
Ábra. 3: A 14,275 percnél eluálódó komponens tömegspektruma.
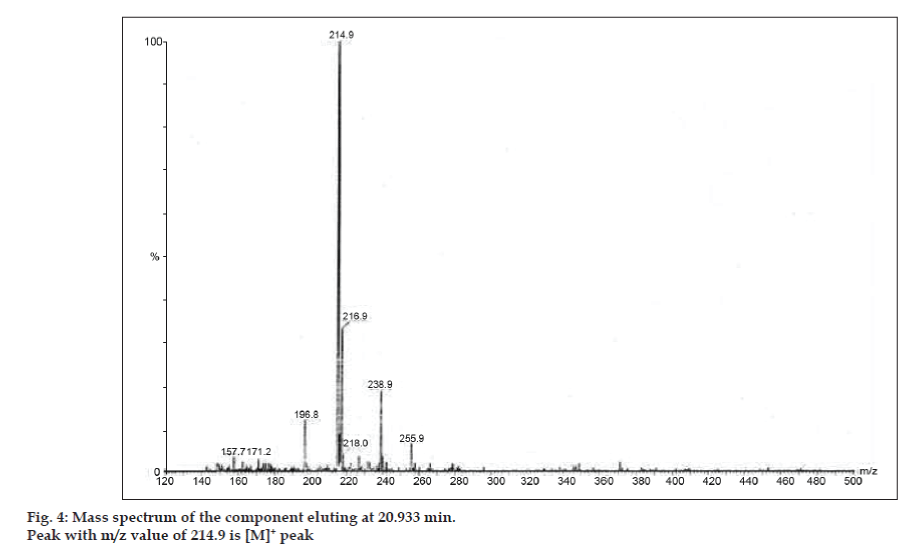
Ábra. 4: A 20,933 percnél eluálódó komponens tömegspektruma.
| Peak no. |
Megmaradási idő (perc) |
Fragmentumionok (m/z) | Identifikáció |
|---|---|---|---|
| 8.324 | 181.9 +, 180.9 +, 150.8 + | Teofillin | |
| 14.275 | 195.7 +, 180.9 +,149.2 +, 137.1+ | Koffein | |
| 20.933 | 216.9 +, 214.9 +, 171.2 +, 157.7 + | A 8- Klórteofillin izomerje |
1. TÁBLÁZAT:
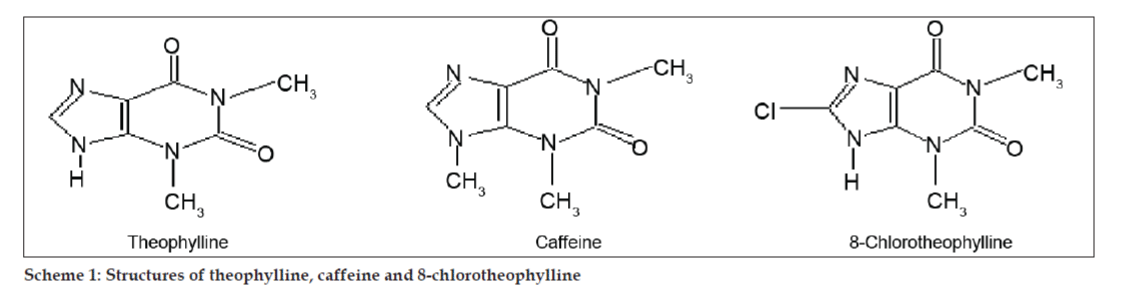
Séma 1: A teofillin, a koffein és a 8-klórtheofillin szerkezete
Köszönet
A szerzők köszönettel tartoznak a Kores (India) Ltd. vállalatnak, Thane-nak a 8-klórteofillin ajándék mintájának biztosításáért.
- Kasture AV, Wadodkar SG, Mahadik KR, More HV. Gyógyszerészeti analízis. Vol. 1. Pune: NiraliPrakashan; 1997. p. 12-4.
- United State Pharmacopoeia, 26. kötet. Rockville, MD: United States Pharmacopoeia Convention, Inc.; 1999. p. 2049-59.
- Ahuja S, Alsante KM. A gyógyszerekben előforduló szennyeződések izolálásának és jellemzésének kézikönyve. California: Academic Press; 2003. p. 6.
- Foye WO, Lemke TL, Williams DA. A gyógyszerkémia alapjai. 4th ed. New Delhi: B. I. Waverly Pvt Ltd; 1995. p. 419.
- Reynolds JE. Martindale-The Extra Pharmacopoeia. 29th ed. London: Pharmaceutical Press; 1989. p. 451,459.
- Wadke DA, Guttman DE. Komplexképződés hatása a reakciósebességre III. Egyes izoalloxazinok kölcsönhatása 8-klórtetofillinnel spektrális, oldhatósági és kinetikai módszerekkel meghatározva. J Pharm Sci 1965;54:1293.
- Bennett MJ, Patchett BP, Worthy E. Egyszerű HPLC módszer az urát meghatározására szérumban és vizeletben 8-klórtotheofillin belső standardként történő felhasználásával. Med Lab Sci 1984;41:108-11.
- Nikolic K, Medenica M. A 8-klórtotheofillin potenciometriás meghatározása. Microchimica Acta 1986;88:5.
- Bebawy LI, El-Kousy NM. Néhány többkomponensű gyógyszerforma egyidejű meghatározása kvantitatív vékonyréteg-kromatográfiás denzitometriás módszerrel. J Pharm Biomed Anal 1999;20:663-70.
- Gil EP, Blazquez LC, Garcia-MoncoCarra RM, Misiego AS. A 8-klórtetofillin polarográfiás viselkedése és meghatározása adagolási formákban. Electroanalysis 1993;5:343.
- Barbas C, Garcia A, Saavedra L, Castro M. A koffein, 8-klórtotheofillin és difenhidramin meghatározására szolgáló módszer optimalizálása és validálása izokratikus nagy teljesítményű folyadékkromatográfiával Stresszvizsgálat a stabilitás értékelésére. J ChromatogrA 2000;870:97-103.
- Kelani KM. Koffein, 8-klórteofillin és klórfenoxamin-hidroklorid egyidejű meghatározása háromkomponensű keverékekben arány-spektroszkópiai nullpontos első derivált spektrofotometriás és kemometriai módszerekkel. J AOAC Int 2005;88:1126-34.