a vörösvértest fő funkcionális alkotóeleme, amely az oxigénszállító fehérje; a hemoprotein egy olyan típusa, amelyben minden molekula egy tetramer, amely négy monomerből áll, amelyeket gyenge kötések tartanak össze. Két pár polipeptidláncból, a globinokból áll, amelyekhez egy-egy vasból és egy protoporfirin molekulából álló hemmolekula kapcsolódik. Jelképe Hb.
A vasatom szabad valenciájú, és képes egy molekula oxigént megkötni. Így minden egyes hemoglobinmolekula egy molekula oxigént képes megkötni. Az oxigénnek az egyik monomer általi megkötése növeli a tetramerben lévő többi monomer oxigén iránti affinitását. Ez teszi a hemoglobint hatékonyabb transzportfehérjévé, mint egy monomer fehérjét, például a mioglobint.
Az oxigénnel kötött hemoglobin (oxi-hemoglobin) élénkpiros színű; az oxigénhez nem kötött hemoglobin (dezoxi-hemoglobin) sötétebb. Ez magyarázza az artériás vér élénkpiros színét, amelyben a hemoglobin körülbelül 97 százalékban telített oxigénnel. A vénás vér sötétebb, mert csak körülbelül 20-70 százalékban telített, attól függően, hogy a szövetek mennyi oxigént használnak fel. A hemoglobin szén-monoxid iránti affinitása 210-szer erősebb, mint az oxigén iránti affinitása. A képződött komplex (karboxi-hemoglobin) nem képes oxigént szállítani. Így a szén-monoxid-mérgezés hipoxiát és fulladást eredményez.
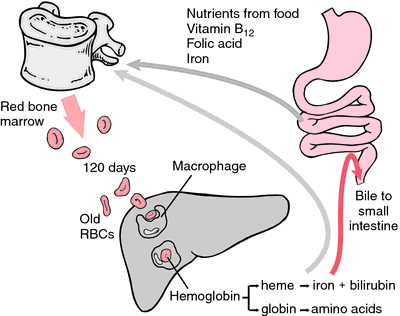
A hemoglobin másik, oxigént szállítani nem képes formája a methemoglobin, amelyben a vasatom a +3 oxidációs állapotba oxidálódott. A vörösvértest 120 napos élettartama alatt a hemoglobin lassan oxidálódik methemoglobinná. Legalább négy különböző enzimrendszer képes a methemoglobint visszafordítani hemoglobinná. Ha ezek hibásak vagy túlterheltek, methemoglobinémia alakulhat ki, és a magas methemoglobinszint nehézlégzést és cianózist okozhat.
A hemoglobin másodlagos funkciója a vér pufferrendszerének része. A globinláncok hisztidinmaradványai gyenge bázisként működnek, hogy minimalizálják a vér pH-értékének változását, amely az oxigén felszívódása és a szén-dioxid felszabadulása során a tüdőben, valamint az oxigén szállítása és a szövetekből történő szén-dioxid felvétele során következik be.
Amint az eritrociták elhasználódnak vagy károsodnak, a retikuloendothelialis rendszer makrofágjai bekebelezik őket. A hem porfiringyűrűje a máj által ürített epefestékké, bilirubinná alakul át. A vas a csontvelőbe kerül, hogy beépüljön az újonnan képződő eritrociták hemoglobinjába.
A vér hemoglobinkoncentrációja a hematokrit függvényében változik. A vér hemoglobinkoncentráció normális értéke férfiaknál 13,5-18,0 g/100 ml, nőknél 12,0-16,0 g/100 ml. A normális átlagos korpuszkuláris hemoglobinkoncentráció, amely a vörösvértesteken belüli koncentráció, 32-36 g/100 ml.
Már számos mutációból eredő kóros hemoglobint fedeztek fel. Néhánynak megváltozott az oxigén-affinitása, néhány instabil, és néhányban a vasatom oxidálódik, ami veleszületett methemoglobinémiát eredményez. Egyes mutációk a hemoglobinszintézis csökkent sebességét eredményezik. Az összes ilyen állapotot hemoglobinopátiának nevezzük.
A leggyakoribb hemoglobinopátia a sarlósejtes betegség, amelyet egy olyan mutáció okoz, amely a β lánc hatodik aminosavát – normális esetben glutaminsavat – valinra cseréli. A hemoglobin α2βS2 változatát Hb S-ként ismerjük. Az egyik lánc csökkent szintézisét eredményező mutációkat thalassémiáknak nevezzük. Ezek eredhetnek az egyik lánc génjének deléciójából vagy a lánc szintézisét szabályozó gén mutációjából.
forrásból.