il principale costituente funzionale del globulo rosso, che serve come proteina portatrice di ossigeno; è un tipo di emoproteina in cui ogni molecola è un tetramero composto da quattro monomeri tenuti insieme da legami deboli. Consiste di due coppie di catene polipeptidiche, le globine, ognuna delle quali ha una molecola di eme attaccata, composta da ferro più una molecola di protoporfirina. Simbolo Hb.
L’atomo di ferro ha una valenza libera e può legare una molecola di ossigeno. Così, ogni molecola di emoglobina può legare una molecola di ossigeno. Il legame dell’ossigeno da parte di un monomero aumenta l’affinità per l’ossigeno degli altri nel tetramero. Questo rende l’emoglobina una proteina di trasporto più efficiente di una proteina monomerica come la mioglobina.
L’emoglobina ossigenata (ossiemoglobina) è di colore rosso vivo; l’emoglobina non legata all’ossigeno (deossiemoglobina) è più scura. Questo spiega il colore rosso vivo del sangue arterioso, in cui l’emoglobina è circa il 97% saturata con ossigeno. Il sangue venoso è più scuro perché è saturo solo dal 20 al 70 per cento, a seconda di quanto ossigeno viene utilizzato dai tessuti. L’affinità dell’emoglobina per il monossido di carbonio è 210 volte più forte della sua affinità per l’ossigeno. Il complesso formato (carbossiemoglobina) non può trasportare ossigeno. Così, l’avvelenamento da monossido di carbonio provoca ipossia e asfissia.
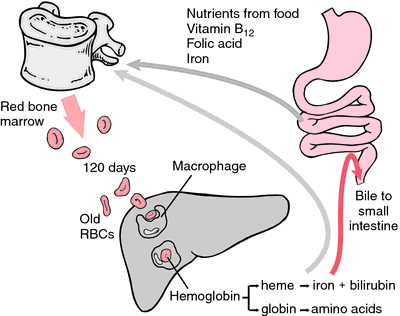
Un’altra forma di emoglobina che non può trasportare ossigeno è la metaemoglobina, in cui l’atomo di ferro è ossidato allo stato di ossidazione +3. Durante i 120 giorni di vita di un globulo rosso, l’emoglobina si ossida lentamente in metemoglobina. Almeno quattro diversi sistemi enzimatici possono riconvertire la metaemoglobina in emoglobina. Quando questi sono difettosi o sovraccarichi, la metemoglobinemia può risultare, con alti livelli di metemoglobina che causano dispnea e cianosi.
Una funzione secondaria dell’emoglobina è come parte del sistema tampone del sangue. I residui di istidina nelle catene globiniche agiscono come basi deboli per minimizzare il cambiamento del pH del sangue che si verifica quando l’ossigeno viene assorbito e l’anidride carbonica rilasciata nei polmoni e quando l’ossigeno viene erogato e l’anidride carbonica assorbita dai tessuti.
Quando gli eritrociti si consumano o vengono danneggiati, vengono ingeriti dai macrofagi del sistema reticoloendoteliale. L’anello porfirinico dell’eme viene convertito nel pigmento biliare bilirubina, che viene escreto dal fegato. Il ferro viene trasportato al midollo osseo per essere incorporato nell’emoglobina degli eritrociti di nuova formazione.
La concentrazione di emoglobina nel sangue varia con l’ematocrito. I valori normali per la concentrazione di emoglobina nel sangue sono da 13,5 a 18,0 g/100 ml nei maschi e da 12,0 a 16,0 g/100 ml nelle femmine. La concentrazione media normale di emoglobina corpuscolare, che è la concentrazione all’interno dei globuli rossi, è da 32 a 36 g/100 ml.
Sono state scoperte molte emoglobine anomale derivanti da mutazioni. Alcune hanno un’affinità di ossigeno alterata, alcune sono instabili, e in alcune l’atomo di ferro è ossidato, con conseguente metemoglobinemia congenita. Alcune mutazioni risultano in un tasso ridotto di sintesi dell’emoglobina. Tutte queste condizioni sono note come emoglobinopatie.
L’emoglobinopatia più comune è la malattia falciforme, causata da una mutazione che sostituisce il sesto aminoacido della catena β, normalmente acido glutammico, con la valina. La variante dell’emoglobina α2βS2 è conosciuta come Hb S. Le mutazioni che risultano in una ridotta sintesi di una delle catene sono chiamate talassemie. Possono derivare dalla delezione del gene per una catena o da una mutazione nel gene regolatore che controlla la sintesi della catena.