Editoriale sul tema della ricerca
The Role of AAA+ Proteins in Riparazione e degradazione delle proteine
Le ATPasi associate a diverse attività cellulari (AAA+) comprendono una superfamiglia di proteine che svolgono una grande varietà di funzioni essenziali alla fisiologia cellulare, compreso il controllo dell’omeostasi proteica, la replicazione del DNA, la ricombinazione, il rimodellamento della cromatina, l’elaborazione dell’RNA ribosomiale, il targeting molecolare, la biogenesi degli organelli e la fusione delle membrane (Hanson e Whiteheart, 2005; Erzberger e Berger, 2006; Snider et al., 2008). I membri di questa superfamiglia sono definiti dalla presenza del cosiddetto dominio AAA+ che contiene i motivi canonici Walker A e B necessari per il legame e l’idrolisi dell’ATP (Hanson e Whiteheart, 2005). In genere, i genomi codificano da circa dieci a diverse centinaia di membri della famiglia AAA+ (Tabella 1; Finn et al., 2017), ognuno dei quali si pensa sia adattato a specifiche nicchie funzionali che necessitano di precisi meccanismi di riconoscimento ed elaborazione del substrato (Hanson e Whiteheart, 2005). La sorprendente radiazione adattativa delle proteine AAA+ per operare in diverse impostazioni illustra l’utilità versatile del dominio AAA+ (Erzberger e Berger, 2006). Le proteine AAA+ formano tipicamente complessi esamerici e agiscono come motori per rimodellare altre proteine, DNA/RNA, o complessi multicomponente (Figura 1). Infatti, molti chaperon e proteasi ATP-dipendenti sono o hanno subunità che appartengono a questa superfamiglia (Figura 1; Olivares et al., 2016).
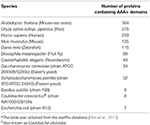
Tabella 1. Numero di proteine AAA+ in organismi modelloa.
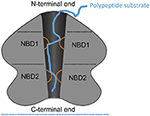
Figura 1. Schema di ClpA come esempio di AAA+ hexamer. Rappresentazione schematica dell’architettura dei domini e delle interazioni di ClpA come esempio di AAA+ hexamer con due domini AAA+ per monomero. Qui, una vista laterale spaccato del ClpA hexamer è mostrato per raffigurare il centrale, polipeptide-conduzione del canale. ClpA contiene tre domini, tra cui un dominio N-terminale e due domini AAA +: nucleotide-binding domain 1 e 2 (NBD1 e NBD2). Il dominio N-terminale interagisce con i regolatori della specificità del substrato, mentre l’estremità C-terminale interagisce con la proteasi a camera ClpP. I loop mobili da NBD1 e NBD2 (arancione) si proiettano nel canale centrale e impegnano il substrato polipeptidico (blu) permettendo così l’accoppiamento dell’idrolisi dell’ATP alla traslocazione del polipeptide attraverso il canale centrale.
Negli ultimi anni, ci sono stati notevoli progressi nell’identificazione della struttura e del meccanismo funzionale di un gran numero di proteine AAA+ (Gates et al., 2017; Puchades et al., 2017; Ripstein et al., 2017; Zehr et al., 2017). In questo tema di ricerca, diversi elementi di questo entusiasmante progresso sono trasmessi in 21 articoli, che comprendono una visione strutturale e meccanicistica dettagliata di diversi chaperon e proteasi AAA+, tra cui: ClpX (Alhuwaider e Dougan; Bittner et al.; Elsholz et al.; LaBreck et al.; Vass et al.), ClpA (Bittner et al.; Duran et al.), ClpB e Hsp104 (Chang et al.Duran et al.; Franke et al.; Johnston et al.), Hsp78 (Abrahão et al.), ClpC (Alhuwaider e Dougan; Elsholz et al.), ClpE (Elsholz et al.), Pontin (Mao e Houry), Reptin (Mao e Houry), FtsH (Alhuwaider e Dougan), proteasoma 19S (Snoberger et al.; Yedidi et al.), Lon (Alhuwaider e Dougan; Bittner et al.; Fishovitz et al.), p97 (Hänzelmann e Schindelin; Saffert et al.; Ye et al.), Pex1/6 (Saffert et al.), CbbQ (Mueller-Cajar), rubisco activase (Bhat et al.), torsine (Chase et al.), e proteasi AAA+ mitocondriali (Glynn). Qui, presentiamo questi affascinanti lavori.
Studi su ClpXP, Lon, e relative proteasi ATP-dipendenti
Nel loro articolo di ricerca, “The Protein Chaperone ClpX Targets Native and Non-native Aggregated Substrates for Remodeling, Disassembly, and Degradation with ClpP,” LaBreck et al. eseguono una serie di eleganti esperimenti per stabilire che ClpX possiede attività di disaggregazione contro polipeptidi che contengono specifici segnali di riconoscimento di ClpX (LaBreck et al.). In presenza di ClpP, ClpX accoppia la disaggregazione di questi substrati alla loro degradazione. Inoltre, essi stabiliscono che ClpXP impedisce l’accumulo di aggregati formati da proteine con segnali di riconoscimento di ClpX in vivo (LaBreck et al.). Questi studi illuminano ClpX come una disaggregazione delle proteine, che era precedentemente sottovalutata.
Nel loro articolo di ricerca, “The Essential Role of ClpXP in Caulobacter crescentus Requires Species Constrained Substrate Specificity”, Vass et al. esplorano funzioni specie-specifiche di ClpX (Vass et al.). Curiosamente, ClpX è essenziale in alcune specie come C. crescentus, ma non essenziale in altri batteri come E. coli (Vass et al.). È importante notare che E. coli ClpX non è stato in grado di integrare C. crescentus ClpX in vivo (Vass et al.). Questa mancanza di attività era dovuta a differenze specie-specifiche nel dominio N-terminale di ClpX, che sono critici per l’elaborazione del DnaX, subunità di carico del morsetto di replicazione in C. crescentus. Quindi, piccole differenze nella specificità di ClpX possono essere particolarmente critiche per specifiche specie batteriche.
Nella loro revisione su “Functional Diversity of AAA+ Protease Complexes in Bacillus subtilis,” Elsholz et al. discutono le funzioni di diverse proteasi AAA+ in B. subtilis, cioè: ClpCP, ClpEP, ClpXP, ClpYQ, LonA/B, e FtsH (Elsholz et al.). Essi discutono come le diverse risposte allo stress controllano la loro espressione e i fenotipi osservati sulla delezione di queste diverse proteasi. La capacità di alcune di queste proteasi di controllare la competenza, la sporulazione, la motilità e la formazione di biofilm sono descritte. Infine, gli autori discutono il targeting di queste proteasi per lo sviluppo di nuovi antibiotici.
Nella loro recensione intitolata “AAA+ Machines of Protein Destruction in Mycobacteria”, Alhuwaider et al. (Alhuwaider e Dougan) discutono i recenti progressi nella determinazione della struttura e della funzione delle AAA+ proteasi dei micobatteri. Queste proteasi sono: ClpXP1P2, ClpC1P1P2, Lon, FtsH, e Mpa. Gli autori discutono anche il sistema Pup-proteasoma (PPS) presente nei micobatteri, che è equivalente al sistema ubiquitina-proteasoma negli eucarioti. Alhuwaider et al. concludono poi con una discussione sui nuovi composti che disregolano o inibiscono l’attività di ClpP1P2 e altri che disregolano ClpC1. Questi composti hanno attività promettenti contro i micobatteri.
In un articolo di ricerca intitolato “The Copper Efflux Regulator CueR Is Subject to ATP-Dependent Proteolysis in Escherichia coli”, Bittner et al. dimostrano che le proteasi AAA+ Lon, ClpXP, e ClpAP sono responsabili della degradazione di E. coli CueR, che è un fattore di trascrizione che controlla l’induzione del sistema Cue di efflusso di rame (Bittner et al.). Gli autori hanno scoperto che il riconoscimento di CueR da parte delle proteasi AAA+ richiede il C-terminale accessibile di CueR. Essi concludono che le proteasi ATP-dipendenti sono necessarie per l’omeostasi del rame in E. coli.
Fishovitz et al. effettuano un confronto dettagliato tra Lon umano ed E. coli nel loro articolo di ricerca intitolato “Utilization of Mechanistic Enzymology to Evaluate the Significance of ADP Binding to Human Lon Protease” (Fishovitz et al.). Usando uno studio meccanicistico dettagliato, hanno trovato che a differenza di E. coli Lon, Lon umano ha una bassa affinità per l’ADP nonostante mostri valori kcat e KM comparabili nell’attività ATPasi. Essi propongono che Lon umano non è regolato da un meccanismo di scambio ADP/ATP promosso dal substrato. Queste differenze tra Lon umano e E. coli potrebbero permettere lo sviluppo futuro di inibitori Lon specie-specifici.
Nella sua recensione su “Multifunctional Mitochondrial AAA Proteases”, il Dr. Glynn discute le due proteasi AAA mitocondriali, i-AAA e m-AAA (Glynn). Entrambe sono proteine di membrana interna mitocondriale. Tuttavia, i-AAA proietta i domini dell’ATPasi e della proteasi nello spazio intermembrana mitocondriale, mentre la proteasi m-AAA proietta i domini catalitici nella matrice. Le strutture di queste proteasi sono discusse così come il loro meccanismo di funzione. Le proteasi possono effettuare la completa degradazione del substrato, ma possono anche scindere solo alcuni substrati come per MrpL32 e Atg32.
ClpB e Hsp104
Nella loro mini-review, “Structural Elements Regulating AAA+ Protein Quality Control Machines,” Chang et al. discutono come il poro loop-1, il motivo di segnalazione inter-subunità, e il motivo di inserzione Pre-Sensor I potrebbero contribuire all’attività di due disaggregasi Hsp100, ClpB batterica e Hsp104 del lievito (Chang et al.). Essi propongono un modello di come questi elementi strutturali potrebbero permettere al ciclo AAA+ ATPasi di essere accoppiato alla traslocazione del substrato attraverso il canale centrale di ClpB e Hsp104. Questo processo di traslocazione del polipeptide è pensato per sostenere come ClpB e Hsp104 estraggono i polipeptidi dalle strutture aggregate (Chang et al.).
Duran et al. forniscono una “Analisi comparativa della struttura e della funzione dei motori AAA+ ClpA, ClpB, e Hsp104: fili comuni e funzioni separate” nella loro recensione (Duran et al.). Essi discutono la capacità di queste tre proteine AAA+ (ClpA, ClpB e Hsp104) di traslocare polipeptidi attraverso i loro complessi esamerici. Tutte queste proteine hanno due domini AAA+ e sono note per dispiegare le proteine. È importante notare che ClpB e Hsp104 sono anche note per funzionare come disaggreganti, mentre ClpA può formare un complesso con la proteasi ClpP. Gli autori evidenziano la necessità di utilizzare metodi cinetici di stato transitorio per esaminare i meccanismi cinetici di queste proteine motore. Descrivono come l’uso di tali metodi ha permesso loro di mostrare che, per esempio, ClpA trasloca polipeptidi a circa 20 aa s-1, mentre in complesso con la proteasi ClpP, il tasso di traslocazione di ClpA è ancora più alto a circa 35 aa s-1. Gli autori discutono anche l’importanza del chaperone Hsp70 nella funzione di ClpB/Hsp104, e l’osservazione della specificità di specie nell’interazione tra Hsp70 e ClpB/Hsp104.
Nel loro articolo di ricerca intitolato “Mutant Analysis Reveals Allosteric Regulation of ClpB Disaggregase”, Franke et al. eseguono analisi mutazionali sulla disaggregasi ClpB di E. coli per caratterizzare la sua regolazione allosterica (Franke et al.). ClpB può essere diviso in un dominio N-terminale e due domini AAA+ separati da una regione elicoidale chiamata dominio M. Gli autori identificano un residuo altamente conservato nel primo dominio AAA+, A328. Il mutante ClpB-A328V è risultato avere un’attività ATPasica molto elevata e ha mostrato una tossicità cellulare. Inaspettatamente, l’alta attività ATPasi di ClpB-A328V era dovuta principalmente al secondo anello AAA+ come valutato dalla spettrometria di massa a scambio di idrogeno amidico. Gli autori concludono che A328 è un residuo cruciale nel controllo dell’idrolisi dell’ATP in entrambi gli anelli AAA+ di ClpB.
Nel loro articolo di ricerca intitolato “Substrate Discrimination by ClpB and Hsp104,” Johnston et al. descrivono le preferenze innate del substrato di ClpB e Hsp104 in assenza dei sistemi chaperone DnaK e Hsp70 (Johnston et al.). Essi mostrano che la specificità del substrato è determinata dal primo dominio AAA+ in ogni proteina. Hanno raggiunto questa conclusione testando i due chaperon per la loro capacità di agire su diversi substrati modello. Hanno anche testato diverse chimere dei due chaperon.
In “Hsp78 (78 kDa Heat Shock Protein), un membro rappresentativo della famiglia AAA trovato nella matrice mitocondriale di Saccharomyces cerevisiae”, Abrahão et al. discutono la struttura e la funzione di Hsp78 (Abrahao et al.). Hsp78 è il paralogo mitocondriale di Hsp104, che funziona nella disaggregazione e riattivazione delle proteine (Abrahao et al.). Curiosamente, Hsp104 e Hsp78 sono stati persi durante la transizione dai protozoi ai metazoi (Abrahao et al.). Tuttavia, Abrahao et al. discutono l’esistenza di ANKCLP, che appare accanto a Hsp78 e Hsp104 nei protozoi e sopravvive alla transizione evolutiva ai metazoi. ANKCLP possiede un dominio AAA+ simile al dominio di legame nucleotidico 2 (NBD2) di Hsp104 e Hsp78, ma è altrimenti altamente divergente. Curiosamente, le mutazioni in ANKCLP causano aciduria 3-metilglutaconica, atrofia cerebrale progressiva, disabilità intellettuale, neutropenia congenita, cataratta e disturbi del movimento negli esseri umani (Abrahao et al.).
p97
Nella loro revisione, “Structure and Function of p97 and Pex1/6 Type II AAA+ Complexes”, Saffert et al. discutono due diversi complessi AAA+ che rimodellano le proteine substrato ubiquitinate (Saffert et al.). Una funzione di p97 è quella di dislocare i substrati ubiquitinati dalla membrana ER al proteasoma durante la degradazione associata all’ER (Saffert et al.). Al contrario, Pex1/Pex6 è un motore eteroesamerico composto da subunità Pex1 e Pex6 alternate, che è essenziale per la biogenesi e la funzione dei perossisomi. Le recenti strutture di microscopia crioelettronica (crio-EM) di p97 e Pex1/6 sono discusse e le differenze strutturali chiave sono evidenziate.
Nella loro recensione intitolata “Un potente ‘estrattore di proteine’ della cellula: struttura e funzione della p97/CDC48 ATPasi”, Ye et al. riassumono le conoscenze attuali della struttura e della funzione di p97 e il suo ruolo in diverse malattie (Ye et al.). p97 ha due domini AAA+ collegati con un breve linker. Ha anche un dominio N-terminale, che media le sue interazioni con diverse proteine adattatrici. Gli autori forniscono una discussione dettagliata della struttura di p97 e dell’effetto dei nucleotidi sulle sue diverse conformazioni. Questi studi si basano sull’uso di tecniche come l’EM, la cristallografia a raggi X e la microscopia a forza atomica ad alta velocità. Gli autori discutono poi le funzioni multicellulari di questa proteina altamente conservata, compresi i suoi ruoli nella degradazione delle proteine associate all’ER (ERAD), la degradazione associata al mitocondrio (MAD) estraendo i polipeptidi dalla membrana esterna mitocondriale, e la degradazione associata al ribosoma (RAD). Infine, Ye et al. forniscono un riassunto delle mutazioni di p97 che portano a diverse malattie umane come IBMPFD (Inclusion Body Myopathy associated with Paget’s disease of the bone and Frontotemporal Dementia)], FALS (sclerosi laterale amiotrofica familiare), CMT2Y (malattia di Charcot-Marie-Tooth, tipo 2Y), paraplegia spastica ereditaria (HSP), malattia di Parkinson (PD), e malattia di Alzheimer (AD).
Nella loro revisione sulla p97 intitolata “L’interazione delle interazioni dei cofattori e delle modifiche post-traslazionali nella regolazione della AAA+ ATPasi p97”, Hänzelmann e Schindelin discutono come diversi cofattori modulano l’attività della p97 ATPasi (Hanzelmann e Schindelin). Essi sottolineano il fatto che la capacità della p97 di essere coinvolta in un gran numero di processi cellulari è dovuta al gran numero di cofattori che interagiscono con questa proteina. Essi delucidano tre diverse classi di cofattori p97, vale a dire: (i) cofattori di reclutamento del substrato come le proteine UBA-UBX e UFD1-NPL4, (ii) cofattori di elaborazione del substrato come le ubiquitine (E3) ligasi e deubiquitinasi (DUBs), e (iii) cofattori regolatori come le proteine UBX, che possono sequestrare o riciclare gli esameri p97. Gli autori discutono anche il ruolo delle modifiche post-traslazionali sull’attività di p97, e sulle sue interazioni con i suoi cofattori e substrati.
Le proteine AAA+ del proteasoma
In “AAA-ATPasi nella degradazione delle proteine”, Yedidi et al. esaminano le attività di Rpt1, Rpt2, Rpt3, Rpt4, Rpt5, e Rpt6, che sono le AAA+ ATPasi del proteasoma eucariotico, così come alcune delle loro parenti batteriche come PAN, Mpa, e VAT (Yedidi et al.). Si concentrano su nuove tecnologie per capire come queste AAA+ ATPasi funzionano traslocando polipeptidi non piegati nella camera proteolitica della proteasi (Yedidi et al.). I cambiamenti conformazionali all’interno dell’anello AAA+ e della camera proteasica adiacente sembrano generare un meccanismo di pompaggio peristaltico per consegnare i substrati alla degradazione (Yedidi et al.).
Nel loro articolo di ricerca, “The Proteasomal ATPases Use a Slow but Highly Processive Strategy to Unfold Proteins,” Snoberger et al. stabiliscono che le proteine proteasomali AAA+ impiegano un meccanismo motore a bassa velocità ma altamente processuale per consegnare i substrati alla cavità proteolitica del proteasoma (Snoberger et al.). Questo meccanismo è in contrasto con ClpX, che utilizza un meccanismo motore ad alta velocità ma meno processuale per consegnare i substrati alla proteasi ClpP per la degradazione. Queste differenze nel meccanismo motore possono essersi evolute in risposta alle diverse richieste della loro specifica clientela.
Rubisco Activases
Nella loro recensione su “Rubisco Activases: AAA+ Chaperones Adapted to Enzyme Repair” Bhat et al. discutono la funzione unica della Rubisco activase (Rca) nel rimodellamento della Rubisco (Bhat et al.). Rca è un chaperone AAA+ altamente conservato negli organismi fotosintetici, dai batteri alle piante superiori. Rubisco è l’enzima Ribulosio-1,5-bisfosfato carbossilato/ossigenasi, che è coinvolto nella fissazione della CO2 atmosferica durante la fotosintesi. È la proteina più abbondante sulla terra ed è l’enzima chiave nella sintesi di tutta la materia organica del pianeta. Tuttavia, Rubisco è un enzima povero ed è facilmente inibito da prodotti collaterali delle sue reazioni catalitiche o da composti sintetizzati da alcune piante in condizioni di scarsa luminosità. Rca funziona per alleviare o “curare” Rubisco da tali inibizioni problematiche. Gli autori discutono la struttura di Rca di diverse specie e i potenziali meccanismi della sua funzione.
Il dottor Mueller-Cajar fornisce una recensione su “Le diverse macchine AAA+ che riparano i siti attivi di Rubisco inibiti” (Mueller-Cajar). Egli discute la presenza di tre classi evolutivamente distinte di Rubisco attivasi (Rcas): (1) verde e (2) rosso-tipo Rcas che si trovano principalmente negli eucarioti fotosintetici della stirpe dei plastidi verdi e rossi, rispettivamente, e (3) CbbQO presente nei batteri chemioautotrofi. Discute l’evoluzione di queste attivasi e il loro potenziale uso in biologia sintetica per migliorare l’attività di Rubisco nelle piante.
Torsin
Nel loro articolo di prospettiva, “Torsin ATPases: Harnessing Dynamic Instability for Function,” Chase et al. discutono le Torsin, che sono anche filogeneticamente correlate a NBD2 di Hsp104 del lievito (Chase et al.). Le torsine sono le uniche ATPasi AAA+ localizzate all’interno dell’ER e dell’involucro nucleare connesso (Chase et al.). Curiosamente, mutazioni in TorsinA causano distonia DYT1, un disturbo neurologico negli esseri umani (Chase et al.). Le torsine mostrano una debole attività ATPasi che viene aumentata attraverso la complementazione del sito attivo dovuta al coassemblaggio con specifici cofattori accessori LAP1 e LULL1 (Chase et al.). Chase et al. suggeriscono che l’assemblaggio e il disassemblaggio dinamico dei complessi Torsin/cofattore giocano ruoli importanti nella loro funzione nel traffico nucleare e nell’assemblaggio del complesso nucleare-poro (Chase et al.).
Pontin e Reptin
Nella loro ampia rassegna su “Il ruolo di Pontin e Reptin nella fisiologia cellulare e nell’eziologia del cancro”, Mao e Houry discutono le molteplici funzioni delle ATPasi AAA+ altamente conservate Pontin e Reptin (Mao e Houry). Queste due proteine funzionano tipicamente insieme come un complesso, ma possono anche funzionare indipendentemente. Gli autori evidenziano i ruoli di Pontin e Reptin nel rimodellamento della cromatina. Discutono anche come Pontin e Reptin modulano le attività trascrizionali di diversi proto-oncogeni come MYC e β-catenina. Mao e Houry chiariscono come Pontin e Reptin sono stati trovati richiesti per l’assemblaggio dei complessi di segnalazione PIKK, così come la telomerasi, il fuso mitotico, la RNA polimerasi II e le snoRNP. Gli autori concludono con una panoramica degli sforzi attuali volti a identificare gli inibitori di Pontin e Reptin da sviluppare come nuovi anti-cancro.
Raccomandazioni conclusive
In conclusione, questa raccolta di 21 articoli evidenzia una serie di importanti aspetti strutturali e meccanicistici delle proteine AAA+ coinvolte nella riparazione e degradazione delle proteine. Siamo entusiasti di vedere come il campo continuerà a svilupparsi durante la rivoluzione crio-EM in corso (Egelman, 2016). Prevediamo che la crio-EM permetterà una comprensione più profonda di come queste affascinanti macchine molecolari operano in diverse situazioni (Gates et al., 2017; Puchades et al., 2017; Ripstein et al., 2017; Zehr et al, 2017).
Contributi degli autori
Tutti gli autori elencati hanno dato un contributo sostanziale, diretto e intellettuale al lavoro e lo hanno approvato per la pubblicazione.
Conflict of Interest Statement
Gli autori dichiarano che la ricerca è stata condotta in assenza di qualsiasi relazione commerciale o finanziaria che possa essere interpretata come un potenziale conflitto di interessi.
Acknowledgments
Il lavoro nel laboratorio di WH in questo settore è finanziato da un Canadian Institutes of Health Research Project Grant (PJT-148564). JS è sostenuto da NIH grant R01GM099836.
Egelman, E. H. (2016). La rivoluzione attuale in Cryo-EM. Biophys. J. 110, 1008-1012. doi: 10.1016/j.bpj.2016.02.001
PubMed Abstract | CrossRef Full Text | Google Scholar
Erzberger, J. P., and Berger, J. M. (2006). Relazioni evolutive e meccanismi strutturali delle proteine AAA+. Annu. Rev. Biophys. Biomol. Struct. 35, 93-114. doi: 10.1146/annurev.biophys.35.040405.101933
PubMed Abstract | CrossRef Full Text | Google Scholar
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro nel 2017 – oltre le annotazioni delle famiglie e dei domini proteici. Nucleic Acids Res. 45, D190-D199. doi: 10.1093/nar/gkw1107
PubMed Abstract | CrossRef Full Text | Google Scholar
Gates, S. N., Yokom, A. L., Lin, J., Jackrel, M. E., Rizo, A. N., Kendsersky, N. M., et al. (2017). Ratchet-like polipeptide meccanismo di traslocazione del AAA + disaggregasi Hsp104. Scienza 357, 273-279. doi: 10.1126/science.aan1052
PubMed Abstract | CrossRef Full Text | Google Scholar
Hanson, P. I., e Whiteheart, S. W. (2005). Proteine AAA+: hanno motore, funzionerà. Nat. Rev. Mol.Cell Biol. 6, 519-529. doi: 10.1038/nrm1684
PubMed Abstract | CrossRef Full Text | Google Scholar
Olivares, A. O., Baker, T. A., and Sauer, R. T. (2016). Approfondimenti meccanicistici nelle proteasi AAA+ batteriche e nelle macchine di rimodellamento delle proteine. Nat. Rev. Microbiol. 14, 33-44. doi: 10.1038/nrmicro.2015.4
PubMed Abstract | CrossRef Full Text | Google Scholar
Puchades, C., Rampello, A. J., Shin, M., Giuliano, C. J., Wiseman, R. L., Glynn, S. E., et al. (2017). Struttura della membrana interna mitocondriale AAA + proteasi YME1 dà comprensione nella lavorazione del substrato. Scienza 358:eaao0464. doi: 10.1126/science.aao0464
PubMed Abstract | CrossRef Full Text | Google Scholar
Ripstein, Z. A., Huang, R., Augustyniak, R., Kay, L. E., e Rubinstein, J. L. (2017). Struttura di un AAA + unfoldase nel processo di unfolding substrato. Elife 6:e25754. doi: 10.7554/eLife.25754
PubMed Abstract | CrossRef Full Text | Google Scholar
Snider, J., Thibault, G., e Houry, W. A. (2008). La superfamiglia AAA+ di proteine funzionalmente diverse. Genome Biol. 9:216. doi: 10.1186/gb-2008-9-4-216
PubMed Abstract | CrossRef Full Text | Google Scholar
Zehr, E., Szyk, A., Piszczek, G., Szczesna, E., Zuo, X., e Roll-Mecak, A. (2017). Le strutture a spirale e ad anello di Katanin fanno luce sul colpo di potenza per la recisione del microtubulo. Nat. Struct. Mol. Biol. 24, 717-725. doi: 10.1038/nsmb.3448
PubMed Abstract | CrossRef Full Text | Google Scholar