SONAL DESAI, ARCHITA PATEL E S. Y. GABHE*
C. U. Shah College of Pharmacy, S. N. D. T. Women’s University, Sir Vithaldas Vidya Vihar, Juhu Tara Road, Santacruz (W), Mumbai – 400 049, India
Autore corrispondente: S. Y. GABHE E-mail:
| Data di accettazione | 16-Gen-2011 |
| Data di revisione | 26-Oct-2010 |
| Data di ricezione | 9-Apr-2009 |
DOI: 10.4103/0250-474X.89762
Abstract
Una semplice cromatografia liquida ad alte prestazioni in fase inversa isocratica è stata utilizzata per separare tre impurità presenti nel campione di 8-clorofillina. La LC-MS è stata utilizzata per la caratterizzazione delle impurità. Sulla base dei dati spettrali di massa, le strutture di queste impurità sono state caratterizzate come 3,7-diidro-1,3-dimetil-1H-purina-2,6-dione (impurità I), 3,7-diidro-1,3,7-trimetil-1H-purina-2,6-dione (impurità II) e isomero di 8-cloro- 1,3-dimetil-2,6(3H,1H)-purinedione (impurità III).
Parole chiave
8-clorofillina, impurità, LC-MS, HPLC a fase inversa, teofillina
Introduzione
L’industria dei farmaci sfusi costituisce la base di tutte le industrie farmaceutiche, poiché è la fonte di ingredienti farmaceutici attivi (API) di qualità specifica. La sfida principale per le industrie di farmaci sfusi è produrre il farmaco finale di qualità e purezza richieste, economicamente. La purezza degli API dipende da diversi fattori come le materie prime, i loro metodi di fabbricazione e il tipo di cristallizzazione o processo di purificazione. Tuttavia, è quasi impossibile ottenere materiali assolutamente puri, poiché le impurità vengono incorporate in essi durante la fabbricazione, la purificazione o la conservazione. Un’impurità è qualsiasi componente di una sostanza farmacologica (esclusa l’acqua) che non è un’entità chimica definita come sostanza farmacologica. Per la sostanza farmacologica prodotta mediante sintesi chimica, ICH classifica le impurità in tre categorie: impurità organiche, impurità inorganiche e solventi residui. Le impurità presenti nella sostanza possono essere tossiche, possono cambiare le proprietà fisiche o chimiche della sostanza, rendendola così inutile dal punto di vista medico. Le impurità possono abbassare la durata di conservazione del prodotto e possono causare difficoltà nella formulazione. Pertanto, non solo il controllo delle impurità ma anche la qualificazione delle impurità è una questione critica per le industrie di farmaci sfusi.
Antitiaminici come il dimenidrinato e il teoclato di prometazina sono farmaci ampiamente utilizzati nel trattamento della cinetosi. La 8-clorofillina, chimicamente 8-cloro-1,3-dimetil-2,6(1H, 3H)- purinedione, è un intermedio che viene utilizzato nella preparazione della forma salina di questi farmaci. È essenziale garantire la purezza e la sicurezza del dimenidrinato e del teoclato di prometazina. Per ottenere ciò, l’8-clorotiofillina deve essere ottenuta con la massima purezza e con un profilo di impurità noto.
Dall’indagine della letteratura risulta che Wadke et al. hanno studiato le interazioni di 9-metilisoalloxazine e 3,9-dimetilisoalloxazine con 8-clorotiofillina. La 8-clorotiofillina è stata usata come standard interno per la determinazione dell’urato con il metodo HPLC. È stato anche determinato potenziometricamente. La determinazione simultanea di clorfenossamina cloridrato, 8-clorotiofillina e caffeina in forma di dosaggio multicomponente è stata condotta con un metodo di cromatografia su strato sottile-densitometrico. Gil et al. hanno studiato il comportamento elettroanalitico della 8-clorofillina mediante voltammetria ciclica e polarografia differenziale a impulsi e hanno determinato il suo contenuto in preparati farmaceutici mediante polarografia differenziale a impulsi. Il metodo RP-HPLC indicante la stabilità è stato sviluppato e convalidato per la 8-clorotiofillina insieme a difenidramina e caffeina. Sono stati sviluppati anche metodi spettrofotometrici e chemiometrici con rapporto-spettro zero-crossing in prima derivazione per la determinazione simultanea di caffeina, 8-cloroteofillina e clorfenossamina cloridrato in miscele ternarie. Finora nessun metodo cromatografico e spettroscopico è stato riportato per la separazione e la caratterizzazione delle impurità presenti nella 8-clorofillina. Pertanto il presente lavoro è stato intrapreso con l’obiettivo di isolare e caratterizzare le impurità presenti in 8-clorofillina utilizzando tecniche analitiche moderne.
8-clorofillina era un campione regalo di Kores (India) Ltd., Thane. Tutti gli altri prodotti chimici e reagenti sono stati acquistati da S. D. Fine Chemicals Ltd (Mumbai, India). I solventi utilizzati per TLC e studi TLC preparativi erano di grado analitico e quelli utilizzati per studi HPLC erano di grado HPLC. L’acetato di sodio triidrato di grado AR è stato utilizzato per la preparazione del buffer.
Inizialmente sono stati eseguiti studi TLC per conoscere il numero di impurità presenti nel campione. Piastre TLC prerivestite di gel di silice 60GF254 (Merck) sono state usate come fase stazionaria. Il campione è stato sciolto in una quantità minima di acetato di etile e questa soluzione è stata utilizzata per la macchiatura delle piastre TLC. Sono state provate varie fasi mobili. Acetato di etile:toluene:acido acetico glaciale (10:0.3:0.5 v/v/v) ha mostrato una migliore separazione rispetto alle altre fasi mobili. Quattro componenti sono stati separati dal campione di 8-clorofillina usando la TLC con i valori Rf di 0,029, 0,132, 0,198 e 0,852, rispettivamente. La 8-clorofillina aveva un Rf di 0,852.
Una volta sviluppata la fase mobile per la TLC, si è cercato di separare la miscela usando la TLC preparatoria. Il campione è stato dissolto in una quantità minima di acetato di etile e macchiato in forma di banda. Tutte e tre le impurità sono state separate dalla 8-clorofillina mediante TLC preparativa utilizzando le stesse condizioni cromatografiche usate negli studi TLC. Le diverse bande sono state raccolte separatamente ed estratte con acetato di etile. Poiché le quantità di ciascuna impurità isolata I, II e III erano molto inferiori, è stato deciso di isolare queste impurità collettivamente. Le impurità I, II e III sono state designate collettivamente come frazione I. La frazione I è stata isolata dalla 8-clorofillina mediante TLC preparativa. Poiché ogni impurità non è stata isolata separatamente, non è stato possibile intraprendere diverse tecniche di identificazione come IR, NMR. Si è deciso di effettuare ulteriori indagini utilizzando la LC-MS, che permette la separazione e la caratterizzazione simultanea. Prima dell’analisi LC-MS, è stato sviluppato un profilo HPLC per la frazione I. Per gli studi HPLC è stato utilizzato un cromatografo liquido ad alte prestazioni Tosoh dotato di pompa alternata CCPM, un controller di pompa PX8010 e un rivelatore UV. Un loop di 20 µl di capacità è stato montato sull’unità della valvola di iniezione. La frazione I è stata sciolta in acetonitrile ed è stata sottoposta ad analisi HPLC in fase inversa con fase mobile composta da acetonitrile: acetato di sodio triidrato (pH 3,57; 0,01 M) (5:95 v/v). La colonna scelta era Phenomenex ODS (250×4.6 mm I.D.; dimensione delle particelle 5 µm). La velocità di flusso era di 1,5 ml/min e la rilevazione è stata monitorata a una lunghezza d’onda di 280 nm. La fase mobile è stata filtrata attraverso un filtro di vetro sinterizzato G5 sotto vuoto prima dell’uso e sonicata per rimuovere le bolle d’aria. L’analisi HPLC della frazione I ha rivelato anche tre picchi con i tempi di ritenzione medi di 8,324 min, 14,275 min e 20,933 min, rispettivamente (fig. 1). Il picco con tempo di ritenzione medio di 2,086 min era il picco del solvente.
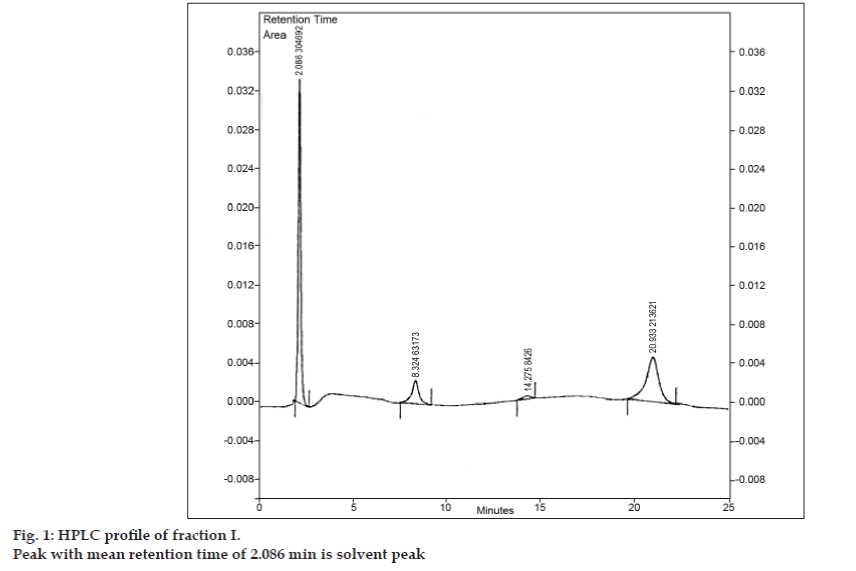
Figura 1: Profilo HPLC della frazione I.
La frazione I è stata quindi sottoposta ad analisi LC-MS per la caratterizzazione delle impurità. Studi LC-MS sono stati effettuati su un sistema in cui parte LC consisteva di serie 1100 HPLC (Agilent Technologies, USA) composto da un degasatore a vuoto (G1322A), pompa quaternaria (G1311A), un campionatore automatico (G1313A) e un rivelatore UV/visibile (G1314A) e parte MS consisteva di spettrometro di massa quadruplo Quattro II (Micromass UK Ltd., UK).
Negli studi LC-MS, le separazioni cromatografiche liquide sono state ottenute mediante colonna Phenomenex C18 (250×4.6 mm, 5 µm) a temperatura ambiente con la fase mobile di acetonitrile: tampone di acetato di sodio (5:95, v/v) ad una velocità di flusso di 1,5 ml/min. Lo spettrometro di massa è stato eseguito in modalità di ionizzazione spray di elettroni positivi (ESI) con rapporto massa/carica (m/z) nell’intervallo di 120-500 m/z. L’azoto è stato utilizzato come gas di nebulizzazione. I dati sono stati acquisiti ed elaborati dal software Masslynx. Sono stati ottenuti i dati spettrali di massa delle impurità (figg. 2, 3 e 4). Il percorso di frammentazione per tre picchi è caratterizzato dalla perdita del gruppo metile e/o del gruppo carbonile. I picchi con valori m/z di 180,9, 195,7 e 214,9 corrispondono rispettivamente ai picchi +, + e +. Secondo i dati MS ottenuti, l’impurità che eluisce a 8,324 min, 14,275 min e 20,933 min erano teofillina (mol. wt. 180), caffeina (mol. wt. 194) e un isomero di 8-clorofillina (mol. wt. 214,5), rispettivamente (Tabella 1). Così, tre impurità sono stati separati e le loro strutture sono stati chiariti sulla base di dati di spettro di massa (Schema 1).
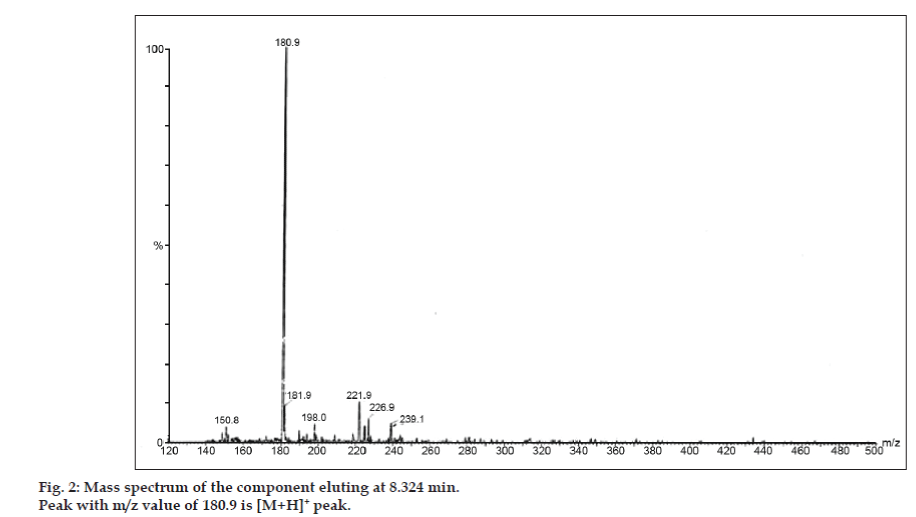
Fig 2: Spettro di massa del componente che eluisce a 8,324 min.
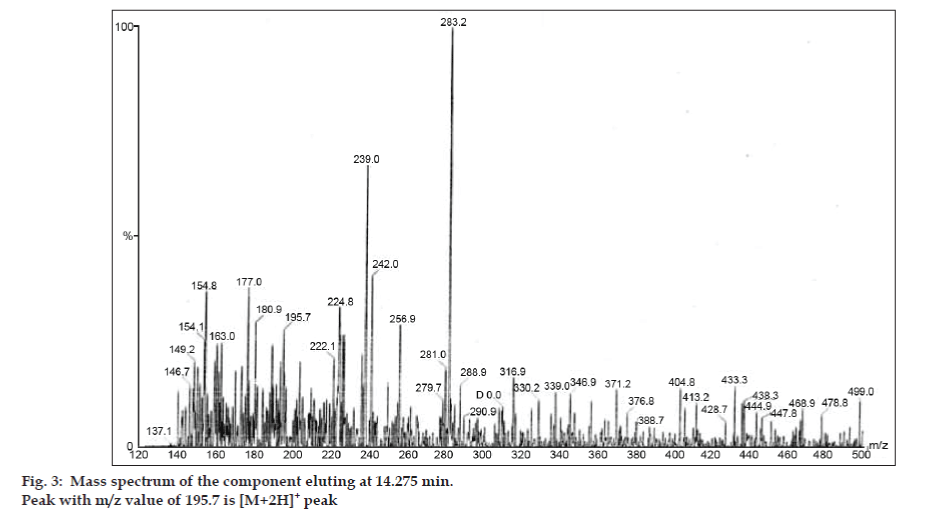
Fig. 3: Spettro di massa del componente che eluisce a 14,275 min.
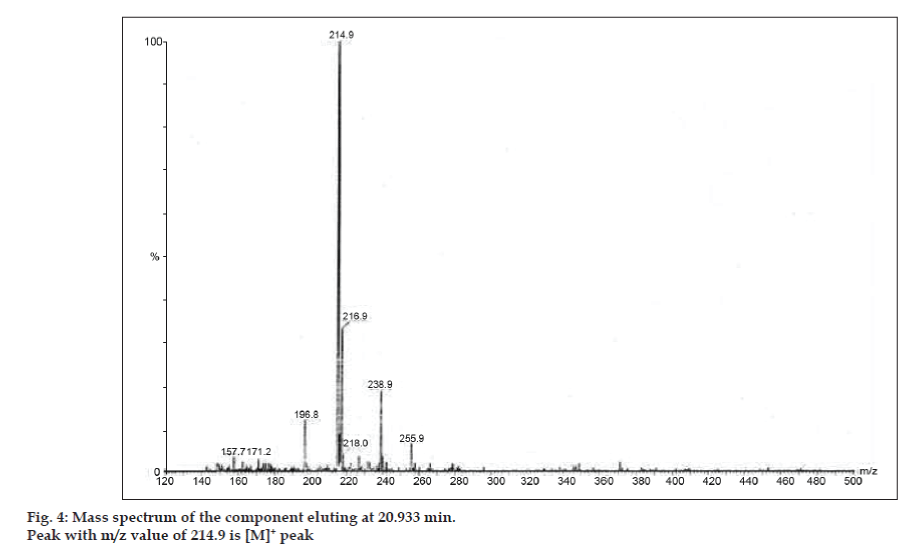
Fig. 4: Spettro di massa del componente che eluisce a 20,933 min.
| Peak no. |
Tempo di ritenzione (min) |
Ioni frammento (m/z) | Identificazione |
|---|---|---|---|
| 8.324 | 181.9 +, 180.9 +, 150.8 + | Teofillina | |
| 14.275 | 195.7 +, 180.9 +,149.2 +, 137.1+ | Caffeina | |
| 20.933 | 216.9 +, 214.9 +, 171.2 +, 157.7 + | Isomero di 8- Clorofillina |
TABELLA 1: IDENTIFICAZIONE HPLC-MS DELLA FRAZIONE I
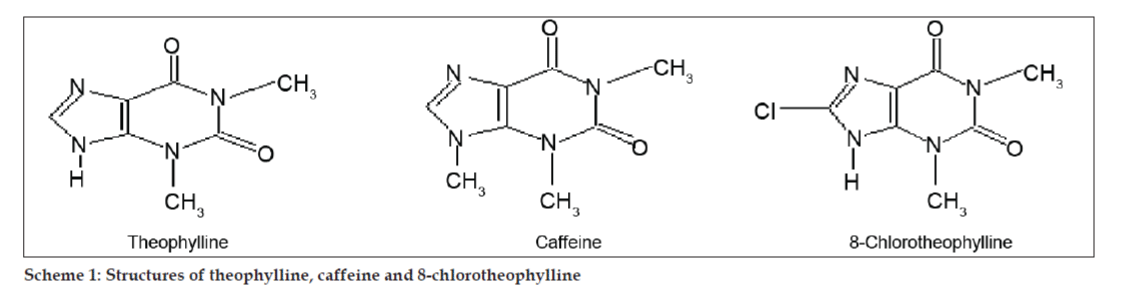
Schema 1: Strutture di teofillina, caffeina e 8-clorofillina
Riconoscimenti
Gli autori sono grati a Kores (India) Ltd., Thane per aver fornito un campione omaggio di 8-clorofillina.
- Kasture AV, Wadodkar SG, Mahadik KR, More HV. Analisi farmaceutica. Vol. 1. Pune: NiraliPrakashan; 1997. p. 12-4.
- Farmacopea degli Stati Uniti, Vol. 26. Rockville, MD: United States Pharmacopoeia Convention, Inc.; 1999. p. 2049-59.
- Ahuja S, Alsante KM. Manuale di isolamento e caratterizzazione delle impurità nei prodotti farmaceutici. California: Academic Press; 2003. p. 6.
- Foye WO, Lemke TL, Williams DA. Principi di chimica medicinale. 4a ed. New Delhi: B. I. Waverly Pvt Ltd; 1995. p. 419.
- Reynolds JE. Martindale-La farmacopea extra. 29° ed. Londra: Pharmaceutical Press; 1989. p. 451,459.
- Wadke DA, Guttman DE. Influenza della formazione di complessi sulla velocità di reazione III. Interazione di alcune isoalloxazine con 8-clorofillina come determinato da metodi spettrali, di solubilità e cinetici. J Pharm Sci 1965;54:1293.
- Bennett MJ, Patchett BP, Worthy E. Un semplice metodo HPLC per la determinazione dell’urato nel siero e nelle urine usando la 8-clorofillina come standard interno. Med Lab Sci 1984;41:108-11.
- Nikolic K, Medenica M. Determinazione potenziometrica della 8-clorotiofillina. Microchimica Acta 1986;88:5.
- Bebawy LI, El-Kousy NM. Determinazione simultanea di alcune forme di dosaggio multicomponente con metodo quantitativo densitometrico di cromatografia su strato sottile. J Pharm Biomed Anal 1999;20:663-70.
- Gil EP, Blazquez LC, Garcia-MoncoCarra RM, Misiego AS. Comportamento polarografico della 8-clorofillina e sua determinazione nelle forme di dosaggio. Elettroanalisi 1993;5:343.
- Barbas C, Garcia A, Saavedra L, Castro M. Ottimizzazione e convalida di un metodo per la determinazione di caffeina, 8-clorofillina e difenidramina mediante cromatografia liquida isocratica ad alte prestazioni Test di stress per la valutazione della stabilità. J ChromatogrA 2000;870:97-103.
- Kelani KM. Determinazione simultanea di caffeina, 8-clorofillina e clorfenossamina cloridrato in miscele ternarie da rapporto-spettro zero-crossing metodi spettrofotometrici e chemiometrici di prima derivazione. J AOAC Int 2005;88:1126-34.