Editorial on the Research Topic
The Role of AAA+ Proteins in Protein Repair and Degradation
ATPazy związane z różnymi czynnościami komórkowymi (AAA+) obejmują nadrodzinę białek, które pełnią wiele różnych funkcji istotnych dla fizjologii komórki, włączając w to kontrolę homeostazy białek, replikację DNA, rekombinację, przebudowę chromatyny, przetwarzanie rybosomalnego RNA, ukierunkowanie molekularne, biogenezę organelli i fuzję błon (Hanson i Whiteheart, 2005; Erzberger i Berger, 2006; Snider i in., 2008). Członków tej nadrodziny definiuje obecność domeny określanej jako AAA+, zawierającej kanoniczne motywy Walkera A i B wymagane do wiązania i hydrolizy ATP (Hanson i Whiteheart, 2005). Typowe genomy kodują od około dziesięciu do kilkuset członków rodziny AAA+ (Tabela 1; Finn i in., 2017), przy czym uważa się, że każdy z nich jest przystosowany do specyficznych nisz funkcjonalnych, wymagających precyzyjnych mechanizmów rozpoznawania i przetwarzania substratów (Hanson i Whiteheart, 2005). Uderzające adaptacyjne promieniowanie białek AAA+ do działania w zróżnicowanych warunkach ilustruje wszechstronną użyteczność domeny AAA+ (Erzberger i Berger, 2006). Białka AAA+ zazwyczaj tworzą kompleksy heksameryczne i działają jak silniki do przebudowy innych białek, DNA/RNA lub kompleksów wieloskładnikowych (Rysunek 1). Rzeczywiście, wiele chaperonów i proteaz zależnych od ATP jest lub ma podjednostki należące do tej nadrodziny (Rysunek 1; Olivares i in., 2016).
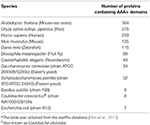
Tabela 1. Liczba białek AAA+ w organizmach modelowychsa.
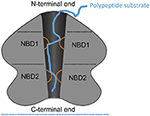
Rysunek 1. Schemat ClpA jako przykład heksameru AAA+. Schematyczne przedstawienie architektury domen i oddziaływań ClpA jako przykładowego heksameru AAA+ z dwiema domenami AAA+ na monomer. W tym miejscu pokazano boczny widok heksameru ClpA w celu zobrazowania centralnego kanału przewodzącego polipeptyd. ClpA zawiera trzy domeny, w tym domenę N-końcową i dwie domeny AAA+: domenę wiążącą nukleotydy 1 i 2 (NBD1 i NBD2). Domena N-końcowa oddziałuje z regulatorami specyficzno¶ci substratowej, podczas gdy koniec C-końcowy oddziałuje z komorow± proteaz± ClpP. Ruchome pętle z NBD1 i NBD2 (pomarańczowe) rzutują do kanału centralnego i angażują substrat polipeptydowy (niebieski) umożliwiając w ten sposób sprzężenie hydrolizy ATP z translokacją polipeptydu przez kanał centralny.
W ciągu ostatnich lat nastąpił znaczny postęp w identyfikacji struktury i mechanizmu funkcjonalnego dużej liczby białek AAA+ (Gates i wsp., 2017; Puchades i wsp., 2017; Ripstein i wsp., 2017; Zehr i wsp., 2017). W tym temacie badawczym, kilka elementów tego ekscytującego postępu jest przekazywanych w 21 artykułach, które obejmują szczegółowe strukturalne i mechanistyczne spojrzenie na kilka chaperonów i proteaz AAA+, w tym: ClpX (Alhuwaider i Dougan; Bittner et al.; Elsholz et al.; LaBreck et al.; Vass et al.), ClpA (Bittner et al.; Duran et al.), ClpB i Hsp104 (Chang et al.; Duran et al.; Franke et al.; Johnston et al.), Hsp78 (Abrahão et al.), ClpC (Alhuwaider i Dougan; Elsholz et al.), ClpE (Elsholz et al.), Pontin (Mao i Houry), Reptin (Mao i Houry), FtsH (Alhuwaider i Dougan), proteasom 19S (Snoberger i wsp.; Yedidi i wsp.), Lon (Alhuwaider i Dougan; Bittner i wsp.; Fishovitz i wsp.), p97 (Hänzelmann i Schindelin; Saffert i in.; Ye i in.), Pex1/6 (Saffert i in.), CbbQ (Mueller-Cajar), aktywator rubisco (Bhat i in.), torsyny (Chase i in.) oraz mitochondrialne proteazy AAA+ (Glynn). Tutaj przedstawiamy te fascynujące prace.
Studies on ClpXP, Lon, and Related ATP-Dependent Proteases
W swoim artykule badawczym, „The Protein Chaperone ClpX Targets Native and Non-native Aggregated Substrates for Remodeling, Disassembly, and Degradation with ClpP,” LaBreck et al. wykonują serię eleganckich eksperymentów w celu ustalenia, że ClpX posiada aktywność dezagregacyjną wobec polipeptydów, które zawierają specyficzne sygnały rozpoznające ClpX (LaBreck i in.). W obecności ClpP, ClpX sprzęga dezagregację tych substratów z ich degradacją. Co ważne, badania te wykazały również, że ClpXP zapobiega gromadzeniu się agregatów tworzonych przez białka posiadające sygnały rozpoznawcze ClpX in vivo (LaBreck i in.). Badania te oświetlają ClpX jako dezagregazę białkową, która wcześniej była niedoceniana.
W swoim artykule badawczym, „The Essential Role of ClpXP in Caulobacter crescentus Requires Species Constrained Substrate Specificity”, Vass i wsp. badają gatunkowo specyficzne funkcje ClpX (Vass i wsp.). Co ciekawe, ClpX jest niezbędna u niektórych gatunków, takich jak C. crescentus, ale nie jest niezbędna u innych bakterii, takich jak E. coli (Vass i in.). Co ważne, E. coli ClpX nie była w stanie uzupełnić C. crescentus ClpX in vivo (Vass et al.). Ten brak aktywności wynikał ze specyficznych dla gatunku różnic w N-końcowej domenie ClpX, które są krytyczne dla przetwarzania podjednostki ładującej klamrę replikacyjną DnaX w C. crescentus. Tak więc niewielkie różnice w specyficzności ClpX mogą być szczególnie krytyczne dla konkretnych gatunków bakterii.
W swoim przeglądzie na temat „Functional Diversity of AAA+ Protease Complexes in Bacillus subtilis”, Elsholz i wsp. omawiają funkcje kilku proteaz AAA+ w B. subtilis, a mianowicie: ClpCP, ClpEP, ClpXP, ClpYQ, LonA/B i FtsH (Elsholz i wsp.). Omówiono, w jaki sposób różne reakcje stresowe kontrolują ich ekspresję i jakie fenotypy obserwuje się po delecji tych różnych proteaz. Opisano zdolność niektórych z tych proteaz do kontrolowania kompetencji, sporulacji, ruchliwości i tworzenia biofilmu. Wreszcie, autorzy omawiają ukierunkowanie tych proteaz na rozwój nowych antybiotyków.
W swoim przeglądzie zatytułowanym „AAA+ Machines of Protein Destruction in Mycobacteria,” Alhuwaider et al. (Alhuwaider i Dougan) omawiają ostatnie postępy w określaniu struktury i funkcji AAA+ proteaz prątków. Proteazy te to: ClpXP1P2, ClpC1P1P2, Lon, FtsH i Mpa. Autorzy omawiają również obecny u prątków system Pup-proteasom (PPS), który jest odpowiednikiem systemu ubikwityna-proteasom u eukariotów. Alhuwaider i wsp. kończą swoją pracę omówieniem nowych związków, które dysregulują lub hamują aktywność ClpP1P2 oraz innych, które dysregulują ClpC1. Związki te mają obiecującą aktywność przeciwko prątkom.
W artykule badawczym zatytułowanym „The Copper Efflux Regulator CueR Is Subject to ATP-Dependent Proteolysis in Escherichia coli”, Bittner i wsp. wykazują, że proteazy AAA+ Lon, ClpXP, i ClpAP są odpowiedzialne za degradację E. coli CueR, który jest czynnikiem transkrypcyjnym kontrolującym indukcję systemu Cue efflux miedzi (Bittner i wsp.). Autorzy stwierdzili, że rozpoznanie CueR przez proteazy AAA+ wymaga dostępnego C-terminusa CueR. Wnioskują oni, że proteazy zależne od ATP są wymagane dla homeostazy miedzi w E. coli.
Fishovitz i wsp. przeprowadzają szczegółowe porównanie między ludzką i E. coli Lon w swoim artykule badawczym zatytułowanym „Utilization of Mechanistic Enzymology to Evaluate the Significance of ADP Binding to Human Lon Protease” (Fishovitz i wsp.). Wykorzystując szczegółowe badania mechanistyczne, stwierdzili oni, że w przeciwieństwie do E. coli Lon, ludzka Lon ma niskie powinowactwo do ADP, mimo że wykazuje porównywalne wartości kcat i KM w aktywności ATPazy. Proponują oni, że ludzka Lon nie jest regulowana przez mechanizm wymiany ADP/ATP promowany przez substrat. Te różnice pomiędzy ludzką i E. coli Lon mogą umożliwić w przyszłości rozwój gatunkowo specyficznych inhibitorów Lon.
W swoim przeglądzie „Multifunctional Mitochondrial AAA Proteases,” Dr. Glynn omawia dwie mitochondrialne proteazy AAA, i-AAA i m-AAA (Glynn). Obie są białkami błony wewnętrznej mitochondrium. Jednakże i-AAA kieruje domeny ATPazy i proteazy do mitochondrialnej przestrzeni międzybłonowej, podczas gdy proteaza m-AAA kieruje domeny katalityczne do macierzy. Omówiono struktury tych proteaz oraz mechanizm ich działania. Proteazy mogą dokonywać całkowitej degradacji substratów, ale także mogą rozszczepiać tylko niektóre substraty, takie jak dla MrpL32 i Atg32.
ClpB i Hsp104
W swoim mini-przeglądzie, „Structural Elements Regulating AAA+ Protein Quality Control Machines,” Chang i wsp. omawiają, jak pętla porowa-1, motyw sygnalizacji między podjednostkami i motyw wstawki Pre-Sensor I mogą przyczyniać się do aktywności dwóch dezagregaz Hsp100, bakteryjnej ClpB i drożdżowej Hsp104 (Chang et al.). Zaproponowali oni model, w jaki sposób te elementy strukturalne mogą umożliwić sprzężenie cyklu ATPazy AAA+ z translokacją substratu przez centralny kanał ClpB i Hsp104. Uważa się, że ten proces translokacji polipeptydów leży u podstaw tego, jak ClpB i Hsp104 wydobywają polipeptydy z zagregowanych struktur (Chang i in.).
Duran i in. przedstawiają „Comparative Analysis of the Structure and Function of AAA+ Motors ClpA, ClpB, and Hsp104: Common Threads and Disparate Functions” w swoim przeglądzie (Duran i in.). Omawiają oni zdolność tych trzech białek AAA+ (ClpA, ClpB i Hsp104) do translokacji polipeptydów poprzez ich heksameryczne kompleksy. Wszystkie te białka mają dwie domeny AAA+ i są znane z rozkładania białek. Co ważne, ClpB i Hsp104 funkcjonują również jako dezagregazy, a ClpA może tworzyć kompleks z proteazą ClpP. Autorzy zwracają uwagę na potrzebę wykorzystania metod kinetyki stanów przejściowych do badania mechanizmów kinetycznych tych białek motorycznych. Opisują, że zastosowanie takich metod pozwoliło im wykazać, że np. ClpA translokacja polipeptydów odbywa się z prędkością ok. 20 aa s-1, natomiast w kompleksie z proteazą ClpP szybkość translokacji ClpA jest jeszcze większa i wynosi ok. 35 aa s-1. Autorzy omawiają również znaczenie chaperonu Hsp70 w funkcji ClpB/Hsp104 oraz obserwację specyficzności gatunkowej w interakcji pomiędzy Hsp70 i ClpB/Hsp104.
W swoim artykule badawczym zatytułowanym „Mutant Analysis Reveals Allosteric Regulation of ClpB Disaggregase,” Franke i wsp. przeprowadzają analizę mutacyjną na dezagregazie E. coli ClpB w celu scharakteryzowania jej regulacji allosterycznej (Franke i wsp.). ClpB można podzielić na domenę N-końcową i dwie domeny AAA+ rozdzielone helikalnym regionem zwanym domeną M. Autorzy identyfikują wysoce konserwowaną resztę w pierwszej domenie AAA+, A328. Stwierdzono, że mutant ClpB-A328V ma bardzo wysoką aktywność ATPazy i wykazuje toksyczność komórkową. Nieoczekiwanie, wysoka aktywność ATPazowa ClpB-A328V była spowodowana głównie przez drugi pierścień AAA+, jak oceniono za pomocą spektrometrii masowej z wymianą wodorową amidów. Autorzy wnioskują, że A328 jest kluczową resztą w kontrolowaniu hydrolizy ATP w obu pierścieniach AAA+ ClpB.
W swoim artykule badawczym zatytułowanym „Substrate Discrimination by ClpB and Hsp104,” Johnston i wsp. opisują wrodzone preferencje substratowe ClpB i Hsp104 przy braku systemów chaperonowych DnaK i Hsp70 (Johnston i wsp.). Wykazują oni, że specyficzność substratowa jest determinowana przez pierwszą domenę AAA+ w każdym z białek. Doszli do tego wniosku testując oba chaperony pod kątem ich zdolności do działania na kilka modelowych substratów. Testowali również różne chimery tych dwóch chaperonów.
W „Hsp78 (78 kDa Heat Shock Protein), a Representative AAA Family Member Found in the Mitochondrial Matrix of Saccharomyces cerevisiae,” Abrahão et al. omawiają strukturę i funkcję Hsp78 (Abrahao et al.). Hsp78 jest mitochondrialnym paralogiem Hsp104, który funkcjonuje w dezagregacji i reaktywacji białek (Abrahao i in.). Co ciekawe, Hsp104 i Hsp78 zostały utracone w momencie przejścia z pierwotniaków do metazoa (Abrahao i in.). Jednakże Abrahao i wsp. omawiają istnienie ANKCLP, który pojawia się obok Hsp78 i Hsp104 u pierwotniaków i przetrwał ewolucyjne przejście do metazoa. ANKCLP posiada domenę AAA+ podobną do domeny 2 wiążącej nukleotydy (NBD2) Hsp104 i Hsp78, ale poza tym jest wysoce rozbieżny. Co intrygujące, mutacje w ANKCLP powodują u ludzi kwasurię 3-metyloglutakonową, postępujący zanik mózgu, niepełnosprawność intelektualną, wrodzoną neutropenię, zaćmę i zaburzenia ruchowe (Abrahao i in.).
p97
W swoim przeglądzie, „Structure and Function of p97 and Pex1/6 Type II AAA+ Complexes,” Saffert i wsp. omawiają dwa różne kompleksy AAA+, które remodelują ubikwitynowane białka substratowe (Saffert i wsp.). Jedną z funkcji p97 jest przemieszczanie ubikwitynowanych substratów z błony ER do proteasomu podczas degradacji związanej z ER (Saffert i in.). Z kolei Pex1/Pex6 jest heteroheksamerycznym motorem składającym się z naprzemiennych podjednostek Pex1 i Pex6, który jest niezbędny do biogenezy i funkcjonowania peroksysomów. W przeglądzie zatytułowanym „A Mighty 'Protein Extractor’ of the Cell: Structure and Function of the p97/CDC48 ATPase”, Ye i wsp. podsumowują aktualną wiedzę na temat struktury i funkcji p97 oraz jego roli w kilku chorobach (Ye i wsp.). p97 ma dwie domeny AAA+ połączone krótkim łącznikiem. Posiada również N-końcową domenę, która pośredniczy w jej interakcjach z różnymi białkami adaptorowymi. Autorzy szczegółowo omawiają strukturę p97 oraz wpływ nukleotydów na jego różne konformacje. Badania te opierają się na wykorzystaniu takich technik jak EM, krystalografia rentgenowska i szybka mikroskopia sił atomowych. Następnie autorzy omawiają wielokomórkowe funkcje tego wysoce konserwatywnego białka, w tym jego rolę w degradacji białek związanych z ERAD (ER-associated protein degradation), degradacji związanej z mitochondriami (MAD) poprzez ekstrakcję polipeptydów z zewnętrznej błony mitochondrialnej oraz degradacji związanej z rybosomami (RAD). Wreszcie, Ye i wsp. przedstawiają podsumowanie mutacji p97 prowadzących do kilku ludzkich chorób, takich jak IBMPFD (Inclusion Body Myopathy associated with Paget’s disease of the bone and Frontotemporal Dementia)], FALS (familial amyotrophic lateral sclerosis), CMT2Y (Charcot-Marie-Tooth disease, type 2Y), dziedziczne spastyczne paraplegie (HSP), choroba Parkinsona (PD) i choroba Alzheimera (AD).
W swoim przeglądzie dotyczącym p97 zatytułowanym „The Interplay of Cofactor Interactions and Post-translational Modifications in the Regulation of the AAA+ ATPase p97”, Hänzelmann i Schindelin omawiają, jak różne kofaktory modulują aktywność ATPazy p97 (Hanzelmann i Schindelin). Podkreślają oni fakt, że zdolność p97 do angażowania się w wiele procesów komórkowych wynika z dużej liczby kofaktorów, które oddziałują z tym białkiem. Wyjaśniają oni trzy różne klasy kofaktorów p97, a mianowicie: (i) kofaktory przyciągające substraty, takie jak białka UBA-UBX i UFD1-NPL4, (ii) kofaktory przetwarzające substraty, takie jak ligazy ubikwitynowe (E3) i deubikwitynazy (DUBs) oraz (iii) kofaktory regulacyjne, takie jak białka UBX, które mogą sekwestrować lub utylizować heksamery p97. Autorzy omawiają również rolę modyfikacji potranslacyjnych na aktywność p97 oraz jego interakcje z kofaktorami i substratami.
AA+ Proteins of the Proteasome
In „AAA-ATPases in Protein Degradation,” Yedidi et al. dokonują przeglądu aktywności Rpt1, Rpt2, Rpt3, Rpt4, Rpt5 i Rpt6, które są ATPazami AAA+ proteasomu eukariotycznego, jak również niektórych z ich bakteryjnych krewnych, takich jak PAN, Mpa i VAT (Yedidi i in.). Skupiają się na nowych technologiach, aby zrozumieć jak te AAA+ ATPazy funkcjonują poprzez translokację rozwiniętych polipeptydów do komory proteolitycznej proteazy (Yedidi i in.). Zmiany konformacyjne w obrębie pierścienia AAA+ i przyległych komór proteazy wydają się generować mechanizm pompowania perystaltycznego w celu dostarczenia substratów do degradacji (Yedidi i in.).
W swoim artykule badawczym, „The Proteasomal ATPases Use a Slow but Highly Processive Strategy to Unfold Proteins,” Snoberger i in. ustalają, że proteasomalne białka AAA+ wykorzystują mechanizm motoryczny o małej prędkości, ale wysoce procesywny, aby dostarczyć substraty do wnęki proteolitycznej proteasomu (Snoberger i in.). Mechanizm ten kontrastuje z ClpX, który wykorzystuje mechanizm motoryczny o dużej szybkości, ale mniejszej procesywności, aby dostarczyć substraty do proteazy ClpP w celu ich degradacji. Te różnice w mechanizmie motorycznym mogły wyewoluować w odpowiedzi na różne wymagania ich specyficznej klienteli.
Aktywazy Rubisco
W swoim przeglądzie na temat „Rubisco Activases: AAA+ Chaperones Adapted to Enzyme Repair” Bhat et al. omawiają unikalną funkcję aktywazy Rubisco (Rca) w przebudowie Rubisco (Bhat et al.). Rca jest chaperonem AAA+, który jest wysoce konserwatywny w organizmach fotosyntetyzujących od bakterii do roślin wyższych. Rubisco jest enzymem karboksylazy/oksygenazy rybulozo-1,5-bisfosforanowej, który bierze udział w wiązaniu atmosferycznego CO2 podczas fotosyntezy. Jest to najobficiej występujące białko na Ziemi i jest kluczowym enzymem w syntezie całej materii organicznej na naszej planecie. Jednakże, Rubisco jest słabym enzymem i jest łatwo hamowany przez produkty uboczne jego reakcji katalitycznych lub przez związki syntetyzowane przez niektóre rośliny w warunkach słabego oświetlenia. Rca funkcjonuje w celu złagodzenia lub „wyleczenia” Rubisco z takich problematycznych inhibicji. Autorzy omawiają strukturę Rca z różnych gatunków i potencjalne mechanizmy jego działania.
Dr Mueller-Cajar przedstawia przegląd „The Diverse AAA+ Machines that Repair Inhibited Rubisco Active Sites” (Mueller-Cajar). Omawia on obecność trzech ewolucyjnie odrębnych klas aktywaz Rubisco (Rcas): (1) Rcas typu zielonego i (2) czerwonego, które występują głównie u eukariotów fotosyntetyzujących z linii plastydów, odpowiednio zielonego i czerwonego, oraz (3) CbbQO występującej u bakterii chemoautotroficznych. Omawia on ewolucję tych aktywaz i ich potencjalne zastosowanie w biologii syntetycznej do zwiększenia aktywności Rubisco u roślin.
Torsyna
W swoim artykule „Torsin ATPases: Harnessing Dynamic Instability for Function”, Chase i wsp. omawiają torsyny, które są również filogenetycznie spokrewnione z NBD2 drożdżowego Hsp104 (Chase i wsp.). Torsyny są jedynymi ATPazami AAA+ zlokalizowanymi wewnątrz ER i połączonymi z otoczką jądrową (Chase i in.). Co intrygujące, mutacje w TorsinA powodują dystonię DYT1, zaburzenie neurologiczne u ludzi (Chase i in.). Torsyny wykazują słabą aktywność ATPazową, która jest zwiększona poprzez komplementację miejsca aktywnego w wyniku koasocjacji ze specyficznymi kofaktorami pomocniczymi LAP1 i LULL1 (Chase i in.). Chase i wsp. sugerują, że dynamiczny montaż i demontaż kompleksów Torsyna/kofaktor odgrywa ważną rolę w ich funkcji w handlu jądrowym i montażu kompleksu jądrowo-porowego (Chase i wsp.).
Pontin i Reptin
W swoim obszernym przeglądzie na temat „Roli Pontin i Reptin w fizjologii komórkowej i etiologii nowotworów”, Mao i Houry omawiają wiele funkcji wysoce konserwowanych ATPaz AAA+ Pontin i Reptin (Mao i Houry). Te dwa białka zazwyczaj funkcjonują razem jako kompleks, ale mogą też funkcjonować niezależnie. Autorzy podkreślają rolę Pontyny i Reptyny w remodelowaniu chromatyny. Omawiają również, w jaki sposób Pontyna i Reptyna modulują aktywność transkrypcyjną kilku proto-onkogenów, takich jak MYC i β-katenina. Mao i Houry wyjaśniają, w jaki sposób okazało się, że pontyna i reptyna są wymagane do składania kompleksów sygnałowych PIKK, a także telomerazy, wrzeciona mitotycznego, polimerazy RNA II i snoRNP. Autorzy kończą przeglądem obecnych wysiłków zmierzających do zidentyfikowania inhibitorów Pontyny i Reptyny, które mają być opracowane jako nowe leki przeciwnowotworowe.
Uwagi końcowe
Podsumowując, ten zbiór 21 artykułów podkreśla szereg ważnych strukturalnych i mechanistycznych aspektów białek AAA+ zaangażowanych w naprawę i degradację białek. Jesteśmy podekscytowani, aby zobaczyć, jak ta dziedzina będzie nadal się rozwijać podczas trwającej rewolucji cryo-EM (Egelman, 2016). Przewidujemy, że cryo-EM umożliwi głębsze zrozumienie, jak te fascynujące molekularne maszyny działają w zróżnicowanych sytuacjach (Gates i in., 2017; Puchades i in., 2017; Ripstein i in., 2017; Zehr i in., 2017).
Author Contributions
Wszyscy wymienieni autorzy wnieśli istotny, bezpośredni i intelektualny wkład w powstanie tej pracy oraz zatwierdzili ją do publikacji.
Oświadczenie o konflikcie interesów
Autorzy oświadczają, że badania zostały przeprowadzone przy braku jakichkolwiek komercyjnych lub finansowych powiązań, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Podziękowania
Praca w laboratorium WH w tym obszarze jest finansowana przez Canadian Institutes of Health Research Project Grant (PJT-148564). JS jest wspierany przez grant NIH R01GM099836.
Egelman, E. H. (2016). The current revolution in Cryo-EM. Biophys. J. 110, 1008-1012. doi: 10.1016/j.bpj.2016.02.001
PubMed Abstract | CrossRef Full Text | Google Scholar
Erzberger, J. P., and Berger, J. M. (2006). Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35, 93-114. doi: 10.1146/annurev.biophys.35.040405.101933
PubMed Abstract | CrossRef Full Text | Google Scholar
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro w 2017 roku – poza rodziną białek i adnotacjami domenowymi. Nucleic Acids Res. 45, D190-D199. doi: 10.1093/nar/gkw1107
PubMed Abstract | CrossRef Full Text | Google Scholar
Gates, S. N., Yokom, A. L., Lin, J., Jackrel, M. E., Rizo, A. N., Kendsersky, N. M., et al. (2017). Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357, 273-279. doi: 10.1126/science.aan1052
PubMed Abstract | CrossRef Full Text | Google Scholar
Hanson, P. I., and Whiteheart, S. W. (2005). AAA+ proteins: have engine, will work. Nat. Rev. Mol.Cell Biol. 6, 519-529. doi: 10.1038/nrm1684
PubMed Abstract | CrossRef Full Text | Google Scholar
Olivares, A. O., Baker, T. A., and Sauer, R. T. (2016). Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol. 14, 33-44. doi: 10.1038/nrmicro.2015.4
PubMed Abstract | CrossRef Full Text | Google Scholar
Puchades, C., Rampello, A. J., Shin, M., Giuliano, C. J., Wiseman, R. L., Glynn, S. E., et al. (2017). Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358:eaao0464. doi: 10.1126/science.aao0464
PubMed Abstract | CrossRef Full Text | Google Scholar
Ripstein, Z. A., Huang, R., Augustyniak, R., Kay, L. E., and Rubinstein, J. L. (2017). Structure of a AAA+ unfoldase in the process of unfolding substrate. Elife 6:e25754. doi: 10.7554/eLife.25754
PubMed Abstract | CrossRef Full Text | Google Scholar
Snider, J., Thibault, G., and Houry, W. A. (2008). The AAA+ superfamily of functionally diverse proteins. Genome Biol. 9:216. doi: 10.1186/gb-2008-9-4-216
PubMed Abstract | CrossRef Full Text | Google Scholar
Zehr, E., Szyk, A., Piszczek, G., Szczesna, E., Zuo, X., and Roll-Mecak, A. (2017). Katanin spiral and ring structures shed light on power stroke for microtubule severing. Nat. Struct. Mol. Biol. 24, 717-725. doi: 10.1038/nsmb.3448
PubMed Abstract | CrossRef Full Text | Google Scholar
.