główny składnik funkcjonalny czerwonej krwinki, służący jako białko przenoszące tlen; jest to rodzaj hemoproteiny, w której każda cząsteczka jest tetramerem składającym się z czterech monomerów utrzymywanych razem przez słabe wiązania. Składa się z dwóch par łańcuchów polipeptydowych, globin, z których każdy ma dołączoną cząsteczkę hemu składającą się z żelaza i cząsteczki protoporfiryny. Symbol Hb.
Atom żelaza ma wolną walencję i może związać jedną cząsteczkę tlenu. Tak więc, każda cząsteczka hemoglobiny może wiązać jedną cząsteczkę tlenu. Wiązanie tlenu przez jeden monomer zwiększa powinowactwo do tlenu pozostałych w tetramerze. To sprawia, że hemoglobina jest bardziej wydajnym białkiem transportowym niż białko monomeryczne, takie jak mioglobina.
Otlenowana hemoglobina (oksyhemoglobina) ma jasnoczerwony kolor; hemoglobina niezwiązana z tlenem (deoksyhemoglobina) jest ciemniejsza. Odpowiada to za jasnoczerwony kolor krwi tętniczej, w której hemoglobina jest w około 97 procentach nasycona tlenem. Krew żylna jest ciemniejsza, ponieważ jest nasycona tylko w około 20-70%, w zależności od tego, jak dużo tlenu jest wykorzystywane przez tkanki. Powinowactwo hemoglobiny do tlenku węgla jest 210 razy silniejsze niż jej powinowactwo do tlenu. Powstały kompleks (karboksyhemoglobina) nie może transportować tlenu. Dlatego zatrucie tlenkiem węgla prowadzi do niedotlenienia i uduszenia.
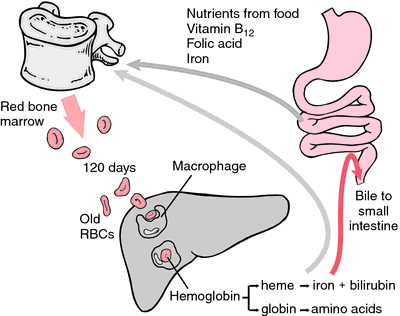
Inną formą hemoglobiny, która nie może transportować tlenu jest methemoglobina, w której atom żelaza jest utleniony do stanu utlenienia +3. Podczas 120-dniowego okresu życia czerwonej krwinki, hemoglobina jest powoli utleniana do methemoglobiny. Co najmniej cztery różne systemy enzymatyczne mogą przekształcić methemoglobinę z powrotem w hemoglobinę. Gdy są one uszkodzone lub przeciążone, może dojść do methemoglobinemii, w której wysoki poziom methemoglobiny powoduje duszność i sinicę.
Drugorzędną funkcją hemoglobiny jest udział w systemie buforowym krwi. Reszty histydynowe w łańcuchach globiny działają jak słabe zasady, aby zminimalizować zmianę pH krwi, która zachodzi w miarę wchłaniania tlenu i uwalniania dwutlenku węgla w płucach oraz dostarczania tlenu i pobierania dwutlenku węgla z tkanek.
Jak erytrocyty zużywają się lub ulegają uszkodzeniu, są połykane przez makrofagi układu siateczkowo-śródbłonkowego. Pierścień porfirynowy hemu jest przekształcany w pigment żółciowy bilirubinę, która jest wydalana przez wątrobę. Żelazo jest transportowane do szpiku kostnego w celu włączenia go do hemoglobiny nowo powstałych erytrocytów.
Stężenie hemoglobiny we krwi zmienia się w zależności od hematokrytu. Normalne wartości stężenia hemoglobiny we krwi wynoszą od 13,5 do 18,0 g/100 ml u mężczyzn i od 12,0 do 16,0 g/100 ml u kobiet. Normalne średnie stężenie hemoglobiny ciałkowej, czyli stężenie wewnątrz czerwonych krwinek, wynosi od 32 do 36 g/100 ml.
Odkryto wiele nieprawidłowych hemoglobin powstałych w wyniku mutacji. Niektóre z nich mają zmienione powinowactwo do tlenu, niektóre są niestabilne, a w niektórych atom żelaza jest utleniony, co powoduje wrodzoną methemoglobinemię. Niektóre mutacje powodują zmniejszenie tempa syntezy hemoglobiny. Wszystkie takie stany są znane jako hemoglobinopatie.
Najczęstszą hemoglobinopatią jest choroba sierpowatokrwinkowa, spowodowana mutacją polegającą na zastąpieniu szóstego aminokwasu w łańcuchu β, normalnie kwasu glutaminowego, waliną. Wariant hemoglobiny α2βS2 znany jest jako Hb S. Mutacje powodujące zmniejszoną syntezę jednego z łańcuchów nazywane są talasemiami. Mogą one wynikać z delecji genu dla danego łańcucha lub z mutacji w genie regulatorowym, który kontroluje syntezę łańcucha.
.