SONAL DESAI, ARCHITA PATEL AND S. Y. GABHE*
C. U. Shah College of Pharmacy, S. N. D. T. Women’s University, Sir Vithaldas Vidya Vihar, Juhu Tara Road, Santacruz (W), Mumbai – 400 049, India
Corresponding Author: S. Y. GABHE E-mail:
| Date of Accepted | 16-Jan-2011 |
| Date of Revised | 26-.Oct-2010 |
| Date of Received | 9-Apr-2009 |
DOI: 10.4103/0250-474X.89762
Abstract
Prostą izokratyczną wysokosprawną chromatografię cieczową w fazie odwróconej zastosowano do rozdzielenia trzech zanieczyszczeń obecnych w próbce 8-chlorotiofyliny. LC-MS został użyty do charakterystyki zanieczyszczeń. Na podstawie danych widma masowego scharakteryzowano struktury tych zanieczyszczeń jako 3,7-dihydro-1,3-dimetylo-1H-puryn-2,6-dion (zanieczyszczenie I), 3,7-dihydro-1,3,7-trimetylo-1H-puryn-2,6-dion (zanieczyszczenie II) i izomer 8-chloro-1,3-dimetylo-2,6(3H,1H)-purynedionu (zanieczyszczenie III).
Słowa kluczowe
8-chlorotiofylina, zanieczyszczenie, LC-MS, HPLC z odwróconą fazą, teofilina
Wprowadzenie
Przemysł leków masowych stanowi podstawę wszystkich gałęzi przemysłu farmaceutycznego, ponieważ jest źródłem aktywnych składników farmaceutycznych (API) o określonej jakości. Głównym wyzwaniem dla przemysłu produkującego leki masowe jest ekonomiczna produkcja leku końcowego o wymaganej jakości i czystości. Czystość API zależy od kilku czynników, takich jak surowce, metody ich wytwarzania oraz rodzaj procesu krystalizacji lub oczyszczania. Jednak uzyskanie absolutnie czystych materiałów jest prawie niemożliwe, ponieważ zanieczyszczenia dostają się do nich podczas produkcji, oczyszczania lub przechowywania. Zanieczyszczenie to każdy składnik substancji leczniczej (z wyłączeniem wody), który nie jest jednostką chemiczną zdefiniowaną jako substancja lecznicza. W przypadku substancji leczniczych produkowanych w drodze syntezy chemicznej ICH klasyfikuje zanieczyszczenia w trzech kategoriach, takich jak zanieczyszczenia organiczne, nieorganiczne i pozostałości rozpuszczalników. Zanieczyszczenia obecne w substancji mogą być toksyczne, mogą zmieniać właściwości fizyczne lub chemiczne substancji, czyniąc ją w ten sposób bezużyteczną medycznie. Zanieczyszczenia mogą obniżać trwałość produktu i powodować trudności w jego formulacji. Dlatego nie tylko kontrola zanieczyszczeń, ale także ich kwalifikacja jest krytycznym zagadnieniem dla przemysłu leków masowych.
Leki przeciwhistaminowe, takie jak dimenhydrynat i teoklat prometazyny są szeroko stosowane w leczeniu choroby lokomocyjnej. 8-chlorotiofylina, chemicznie 8-chloro-1,3-dimetylo-2,6(1H, 3H)- purynedion, jest półproduktem, który jest stosowany w przygotowywaniu postaci soli tych leków. Istotne jest zapewnienie czystości i bezpieczeństwa dimenhydrynatu i teoklanu prometazyny. Aby to osiągnąć, 8-chlorotiofylina musi być otrzymywana z najwyższą czystością i o znanym profilu zanieczyszczeń.
Z przeglądu literatury wynika, że Wadke i wsp. badali interakcje 9-metyloizoaloksazyny i 3,9-dimetyloizoaloksazyny z 8-chlorotiofyliną. 8-chlorotiofilina została użyta jako wzorzec wewnętrzny do oznaczania moczanów metodą HPLC. Oznaczano ją również potencjometrycznie. Jednoczesne oznaczanie chlorowodorku chlorfenoksaminy, 8-chlorotiofyliny i kofeiny w wieloskładnikowej postaci leku przeprowadzono metodą chromatografii cienkowarstwowej-densytometrycznej. Gil i wsp. badali zachowanie elektroanalityczne 8-chlorotiofyliny metodą woltamperometrii cyklicznej i różnicowej polarografii pulsacyjnej oraz oznaczali jej zawartość w preparatach farmaceutycznych metodą różnicowej polarografii pulsacyjnej. Opracowano i zwalidowano metodę RP-HPLC wskazującą na stabilność 8-chlorotiofyliny wraz z difenhydraminą i kofeiną. Opracowano również metody spektrofotometryczne i chemometryczne z zastosowaniem metody ratio-spectra zero-crossing first-derivative do jednoczesnego oznaczania kofeiny, 8-chlorotiofyliny i chlorowodorku chlorfenoksaminy w mieszaninach trójskładnikowych. Dotychczas nie opracowano chromatograficznej i spektroskopowej metody rozdziału i charakterystyki zanieczyszczeń obecnych w 8-chlorotiofylinie. Dlatego obecna praca została podjęta w celu wyizolowania i scharakteryzowania zanieczyszczeń obecnych w 8-chlorotheophylline przy użyciu nowoczesnych technik analitycznych.
8-Chlorotheophylline była próbka dar od Kores (Indie) Ltd., Thane. Wszystkie inne substancje chemiczne i odczynniki pochodziły od S. D. Fine Chemicals Ltd (Mumbai, Indie). Rozpuszczalniki użyte do badań TLC i preparatywnej TLC były klasy analitycznej, a te użyte do badań HPLC były klasy HPLC. Sodium acetate trihydrate of AR grade was used for preparation of buffer.
Initially TLC studies were carried out in order to know the number of impurities present in the sample. Jako fazę stacjonarną zastosowano wstępnie powlekane płytki TLC z żelem krzemionkowym 60GF254 (Merck). Próbka została rozpuszczona w minimalnej ilości octanu etylu i roztwór ten został użyty do nakrapiania płytek TLC. Wypróbowano różne fazy ruchome. Octan etylu:toluen:lodowaty kwas octowy (10:0,3:0,5 v/v/v) wykazał lepszą separację w porównaniu z innymi fazami ruchomymi. Cztery składniki zostały oddzielone od próbki 8-chlorotiofyliny za pomocą TLC z wartościami Rf wynoszącymi odpowiednio 0,029, 0,132, 0,198 i 0,852. 8-chlorotiofylina miała Rf równy 0,852.
Po opracowaniu fazy ruchomej do TLC podjęto próbę rozdzielenia mieszaniny za pomocą preparatywnej TLC. Próbka została rozpuszczona w minimalnej ilości octanu etylu i nakrapiana w postaci pasma. Wszystkie trzy zanieczyszczenia oddzielono od 8-chlorotiofyliny metodą preparatywnej TLC, stosując te same warunki chromatograficzne, jakie zastosowano w badaniach TLC. Poszczególne pasma zostały zebrane oddzielnie i poddane ekstrakcji przy użyciu octanu etylu. Ponieważ ilości każdego wyizolowanego zanieczyszczenia I, II i III były bardzo małe, postanowiono wyizolować te zanieczyszczenia zbiorczo. Zanieczyszczenia I, II i III oznaczono zbiorczo jako frakcję I. Frakcję I wyizolowano z 8-chlorotiofyliny metodą preparatywnej TLC. Ponieważ każde zanieczyszczenie nie zostało wyizolowane oddzielnie, nie można było zastosować różnych technik identyfikacji, takich jak IR i NMR. Zdecydowano się na przeprowadzenie dalszych badań przy użyciu LC-MS, która pozwala na jednoczesne rozdzielenie i scharakteryzowanie zanieczyszczeń. Przed przystąpieniem do analizy LC-MS opracowano profil HPLC dla frakcji I. Do badań HPLC wykorzystano wysokosprawny chromatograf cieczowy firmy Tosoh wyposażony w pompę tłokową CCPM, sterownik pompy PX8010 oraz detektor UV. Do zespołu zaworu iniekcyjnego zamocowano pętlę o pojemności 20 µl. Frakcję I rozpuszczano w acetonitrylu i poddawano analizie HPLC z fazą odwróconą, w której fazę ruchomą stanowił acetonitryl: trójwodny octan sodu (pH 3,57; 0,01 M) (5:95 v/v). Wybrano kolumnę Phenomenex ODS (250×4,6 mm I.D.; wielkość cząstek 5 µm). Szybkość przepływu wynosiła 1,5 ml/min, a detekcja była monitorowana przy długości fali 280 nm. Faza ruchoma była przed użyciem filtrowana przez filtr ze szkła spiekanego G5 w warunkach próżni i poddawana sonikacji w celu usunięcia pęcherzyków powietrza. Analiza HPLC frakcji I ujawniła trzy piki o średnich czasach retencji wynoszących odpowiednio 8,324 min, 14,275 min i 20,933 min (rys. 1). Pik o średnim czasie retencji 2,086 min był pikiem rozpuszczalnika.
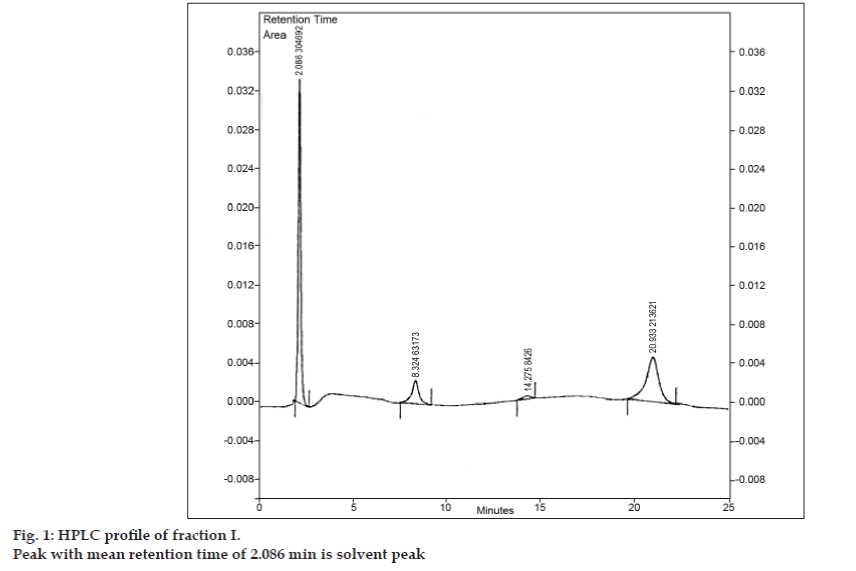
Figura 1: Profil HPLC frakcji I.
Frakcja I została następnie poddana analizie LC-MS w celu scharakteryzowania zanieczyszczeń. Badania LC-MS przeprowadzono w systemie, w którym część LC składała się z HPLC serii 1100 (Agilent Technologies, USA) zawierającego degazer próżniowy (G1322A), pompę czwartorzędową (G1311A), automatyczny próbnik (G1313A) i detektor UV/widoczny (G1314A), a część MS składała się z potrójnego kwadrupolowego spektrometru mas Quattro II (Micromass UK Ltd., UK).
W badaniach LC-MS, rozdziały chromatograficzne cieczowe uzyskano za pomocą kolumny Phenomenex C18 (250×4,6 mm, 5 µm) w temperaturze pokojowej z fazą ruchomą acetonitryl: bufor octanu sodu (5:95, v/v) przy prędkości przepływu 1,5 ml/min. Spektrometr mas pracował w trybie dodatniej jonizacji elektronowej (ESI) ze stosunkiem masy do ładunku (m/z) w zakresie 120-500 m/z. Jako gazu nebulizującego użyto azotu. Dane były gromadzone i przetwarzane przez oprogramowanie Masslynx. Uzyskano widma masowe zanieczyszczeń (rys. 2, 3 i 4). Ścieżka fragmentacji dla trzech pików charakteryzuje się utratą grupy metylowej i/lub grupy karbonylowej. Piki o wartościach m/z 180,9, 195,7 i 214,9 odpowiadają odpowiednio pikom +, + i +. Zgodnie z uzyskanymi danymi MS, zanieczyszczeniami eluującymi w 8,324 min, 14,275 min i 20,933 min były teofilina (mol. mas. 180), kofeina (mol. mas. 194) i izomer 8-chlorotiofyliny (mol. mas. 214,5), odpowiednio (Tabela 1). Tak więc, trzy zanieczyszczenia zostały oddzielone i ich struktury zostały wyjaśnione w oparciu o dane widma masowego (Schemat 1).
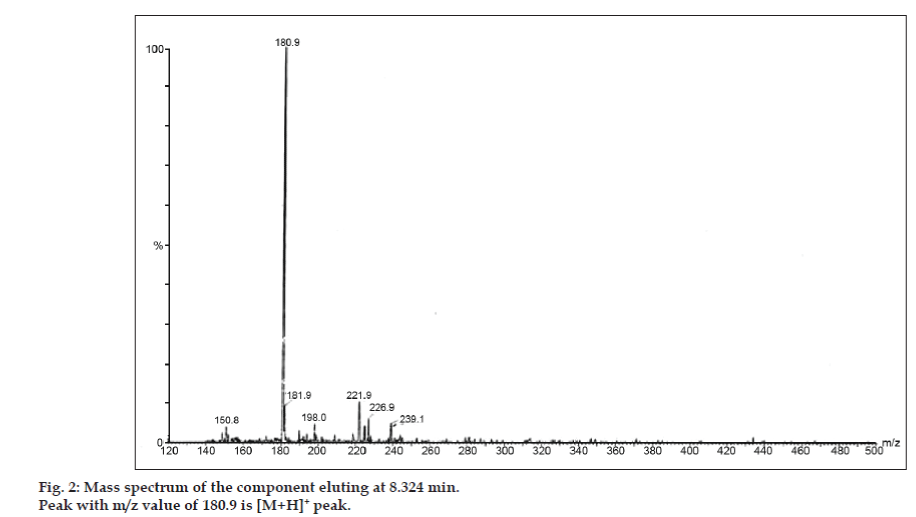
Fig. 2: Widmo masowe składnika eluującego w 8.324 min.
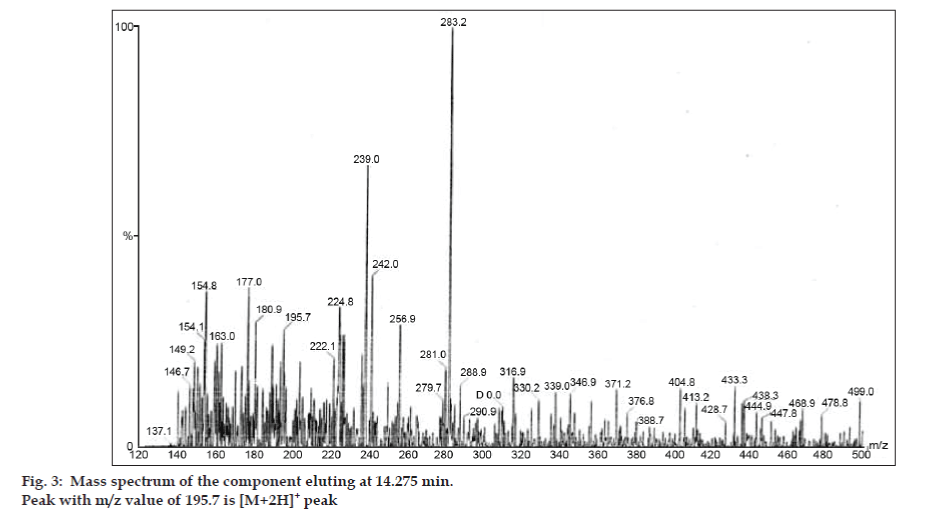
Fig. 3: Widmo masowe składnika eluującego w 14.275 min.
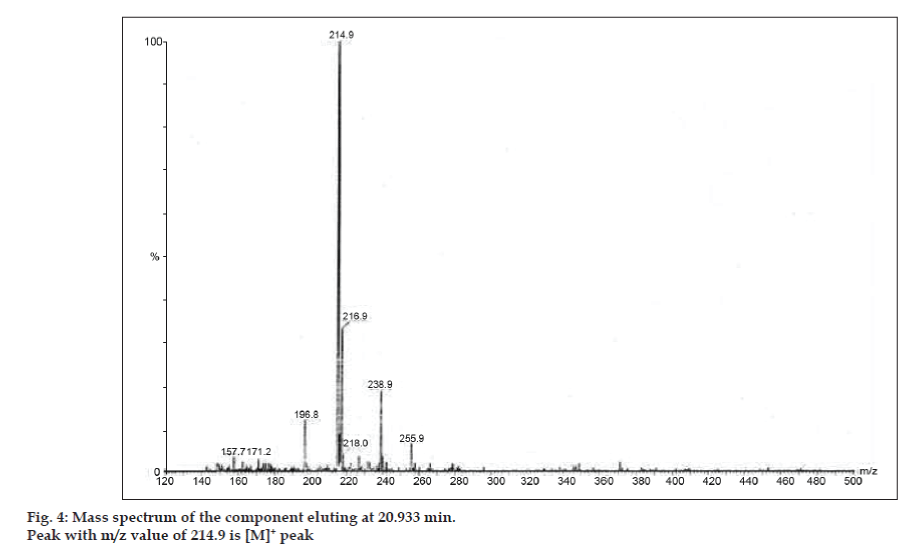
Fig. 4: Widmo masowe składnika eluującego w 20,933 min.
| Peak no. |
Retention time (min) |
Fragment ions (m/z) | Identification |
|---|---|---|---|
| 8.324 | 181.9 +, 180.9 +, 150.8 + | Theophylline | |
| 14.275 | 195.7 +, 180.9 +,149.2 +, 137.1+ | Kofeina | |
| 20.933 | 216.9 +, 214.9 +, 171.2 +, 157.7 + | Isomer 8- chlorotiofyliny |
TABELA 1: HPLC-MS IDENTYFIKACJA FRACTION I
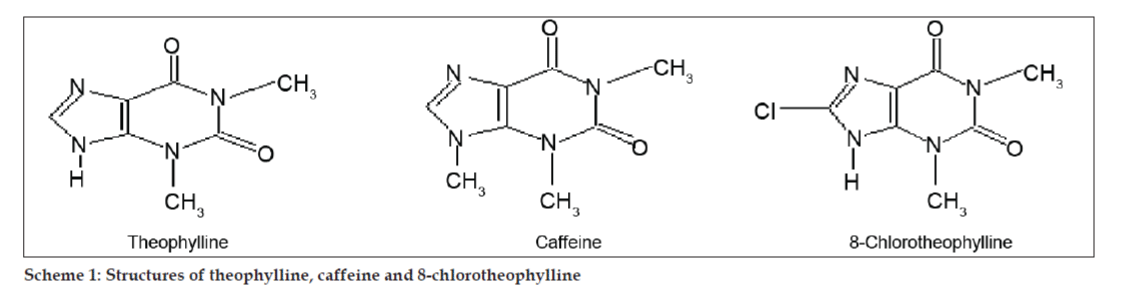
Schemat 1: Struktury teofiliny, kofeiny i 8-chlorotiofyliny
Podziękowania
Autorzy są wdzięczni firmie Kores (India) Ltd., Thane za dostarczenie próbki 8-chlorotiofyliny.
- Kasture AV, Wadodkar SG, Mahadik KR, More HV. Pharmaceutical Analysis. Vol. 1. Pune: NiraliPrakashan; 1997. s. 12-4.
- United State Pharmacopoeia, Vol. 26. Rockville, MD: United States Pharmacopoeia Convention, Inc; 1999. s. 2049-59.
- Ahuja S, Alsante KM. Handbook of isolation and characterization of impurities in pharmaceuticals. California: Academic Press; 2003. s. 6.
- Foye WO, Lemke TL, Williams DA. Principles of Medicinal Chemistry. 4th ed. New Delhi: B. I. Waverly Pvt Ltd; 1995. s. 419.
- Reynolds JE. Martindale-The Extra Pharmacopoeia. 29th ed. London: Pharmaceutical Press; 1989. s. 451,459.
- Wadke DA, Guttman DE. Wpływ tworzenia kompleksów na szybkość reakcji III. Interaction of some isoalloxazines with 8-chlorotheophylline as determined by spectral, solubility, and kinetic methods. J Pharm Sci 1965;54:1293.
- Bennett MJ, Patchett BP, Worthy E. A simple HPLC method for the determination of urate in serum and urine using 8-chlorotheophylline as internal standard. Med Lab Sci 1984;41:108-11.
- Nikolic K, Medenica M. Potencjometryczne oznaczanie 8-chlorotiofyliny. Microchimica Acta 1986;88:5.
- Bebawy LI, El-Kousy NM. Simultaneous determination of some multicomponent dosage forms by quantitative thin layer chromatography densitometric method. J Pharm Biomed Anal 1999;20:663-70.
- Gil EP, Blazquez LC, Garcia-MoncoCarra RM, Misiego AS. Polarographic Behavior of 8-Chlorotheophylline and its Determination in Dosage Forms. Electroanalysis 1993;5:343.
- Barbas C, Garcia A, Saavedra L, Castro M. Optimization and validation of a method for the determination of caffeine, 8-chlorotheophylline and diphenhydramine by isocratic high-performance liquid chromatography Stress test for stability evaluation. J ChromatogrA 2000;870:97-103.
- Kelani KM. Simultaneous determination of caffeine, 8- chlorotheophylline, and chlorphenoxamine hydrochloride in ternary mixtures by ratio-spectra zero-crossing first-derivative spectrophotometric methods and chemometric methods. J AOAC Int 2005;88:1126-34.
.