Wprowadzenie
Reumatoidalne zapalenie stawów (RZS) jest chorobą autoimmunologiczną, o charakterze poligenicznym, charakteryzującą się wielostawowym zapaleniem stawów z objawami układowymi oraz zwiększoną i ciężką chorobowością.1,2 RZS dotyka 0,5%-1% populacji, powodując obniżenie jakości życia, znaczną niepełnosprawność fizyczną i znaczne koszty ekonomiczne.3-6 Kliniczna ekspresja choroby jest zróżnicowana, od łagodnych postaci samoograniczających się do bardzo agresywnej, szybkiej ewolucji, która kończy się zniszczeniem chorego stawu i wynikającą z tego niepełnosprawnością.7
Badania genetyczne potwierdziły istnienie podłoża genetycznego, częściowo związanego z pewnymi genami kodującymi białka biorące udział w odpowiedzi komórek T.1 Odkrycia te wzmacniają znaczenie roli przypisywanej komórkom T w inicjowaniu i utrwalaniu nieprawidłowej odpowiedzi immunologicznej w tej chorobie.8
Patogeneza RZS jest złożona i obejmuje różne populacje komórek związanych z wrodzoną i adaptacyjną odpowiedzią immunologiczną. W patogenezie biorą udział komórki rezydujące w maziówce, takie jak fibroblastyczne synowiocyty B lub makrofagi błony maziowej, oraz komórki zapalne z krwi, takie jak limfocyty T, limfocyty B i monocyty9. Wszystkie one przyczyniają się do agresywnej transformacji fenotypu synowiocytów B i rozwoju intensywnego nacieku zapalnego, którego końcowym efektem jest destrukcja chrząstki i kości podchrzęstnej10,11 (ryc. 1).
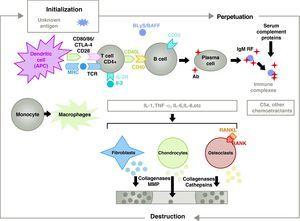
Patofizjologia reumatoidalnego zapalenia stawów. Ogólna organizacja patofizjologiczna reumatoidalnego zapalenia stawów. AC, przeciwciało; BAFF, czynnik aktywujący komórki B; BLyS, stymulator limfocytów B; CD, klaster różnicowania; CPA, komórka prezentująca antygen; CPH, MHC; CTLA4, antygen związany z limfocytami 4 T cytotoksyczna frakcja C5a dopełniacza 5a, FR, czynnik reumatoidalny; Ig, immunoglobulina; IL, interleukina; MMP, metaloproteinazy macierzy; RANK, aktywator receptora czynnika jądrowego B kappa; RANKL, ligand aktywatora receptora dla czynnika jądrowego B kappa; RCT, receptor komórek T; TNF, czynnik martwicy nowotworów.
Obecne leczenie RZS opiera się na podawaniu leków przeciwreumatycznych modyfikujących przebieg choroby (DMARDs) stosowanych samodzielnie lub w skojarzeniu.12. Leki te spowalniają destrukcję stawów, czyli są w stanie modyfikować naturalny przebieg choroby.4,13 Jednak odsetek pacjentów, u których uzyskuje się zadowalającą odpowiedź kliniczną jest niski i często u dużego odsetka chorych wymaga dodania leku biologicznego.9,13-15
W ostatnich latach zidentyfikowano nowe cząsteczki i cele terapeutyczne, których blokada mogłaby zmniejszyć lub wyeliminować przewlekłą odpowiedź zapalną. Jedną z tych nowych cząsteczek jest abatacept. Abatacept jest w pełni humanizowaną konstrukcją białkową, składającą się z zewnątrzkomórkowej domeny ludzkiego antygenu 4 związanego z limfocytami T cytotoksycznymi (CTL4) i genetycznie zmodyfikowanego fragmentu regionu Fc ludzkiej immunoglobuliny G1 (IgG1), która hamuje kostymulację komórek T działających na prawdziwe jądro odpowiedzi immunologicznej, a zatem na początku choroby.
Aktywacja komórek T
Efektywna aktywacja immunologiczna komórek T wymaga udziału dwóch grup receptorów błonowych na komórkach prezentujących antygen (APC)14 (ryc. 1 i 2). Pierwsza z nich jest nośnikiem, za pomocą którego APC dostarczają do komórki T uprzednio przetworzony specyficzny antygen. Pomimo ogromnego wysiłku włożonego w te badania, nadal nie udało się zidentyfikować antygenów artretygenowych wywołujących RZS.8 Prezentacja przez APC antygenu, przeciwko któremu powstaje swoista odpowiedź immunologiczna, jest zorganizowana poprzez kompleks trójcząsteczkowy obejmujący: cząsteczki głównego kompleksu zgodności tkankowej (MHC) obecne w APC, antygen, przeciwko któremu rozwija się odpowiedź immunologiczna, oraz receptor błonowy na komórce T (TCR) swoisty dla tego antygenu15 (szlak sygnałowy lub sygnalizacyjny odpowiedzi immunologicznej 1).
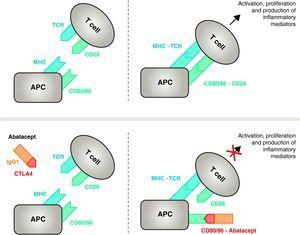
Mechanizm działania abataceptu. Fragment abataceptu obejmujący zewnątrzkomórkową domenę CTLA4 wiąże się z receptorami CD80/CD86, uniemożliwiając lub wypierając jego interakcję z receptorem CD28. W ten sposób selektywnie blokuje specyficzne wiązanie CD80/CD86 z receptorem CD28, co stanowi, patofizjologicznie, blokowanie drugiego sygnału aktywacji immunologicznej, a tym samym aktywacji komórek T CPA, komórka prezentująca antygen; MHC, główny kompleks zgodności tkankowej; TCR, receptor komórek T.
Aby zahamować pełną aktywację, komórki T wymagają drugiego zestawu receptorów komunikacji międzykomórkowej między APC a komórkami T, która zachodzi poprzez szlaki kostymulacyjne i stanowi tak zwany 2 sygnał odpowiedzi immunologicznej.14 Chociaż istnieje kilka szlaków kostymulacyjnych, jeden z nich jest zasadniczy – wiązanie receptorów CD80 (B7-1)/CD86 (B7-2) na błonie CPA z receptorem CD28 na limfocytach T.10,16 Jednoczesna aktywacja obu wyzwala intensywną sygnalizację wewnątrzkomórkową w limfocytach T, niezbędną do pełnej aktywacji, proliferacji, przeżycia i produkcji cytokin.8. W 24-48h od aktywacji limfocytów T, ta sama sygnalizacja wewnątrzkomórkowa inicjuje mechanizm regulacyjny, który ma na celu dezaktywację samej odpowiedzi. Indukuje to ekspresję CTLA411 na błonie komórkowej limfocytów, którego zadaniem jest konkurowanie z CD28 ze względu na większe powinowactwo wiązania do CD80/CD86.17,18
Aktywacja obu podgrup limfocytów T, CD4+ i CD8+ zależy od receptora kostymulującego CD28. Komórki T CD4+ są komórkami T pomocniczymi. Rozpoznają one peptydy prezentowane przez cząsteczki MHC klasy II obecne na APC. Antygeny te pochodzą ze szlaku egzogennego, który przetwarza patogeny, takie jak bakterie. Wiele chorób autoimmunologicznych jest związanych z patologiczną odpowiedzią limfocytów T CD4+. Komórki T CD8+ są z kolei limfocytami cytotoksycznymi (CTL). Komórki T CD8+ rozpoznają antygeny, głównie wirusowe i nowotworowe, prezentowane przez cząsteczki MHC klasy I. Po aktywacji komórki CD8+ pośredniczą w niszczeniu komórek docelowych poprzez produkcję perforyny, granzymów i interferonu (IFN)-g. Oba podtypy limfocytów T są aktywowane przez kostymulację z CD2815, chociaż aktywacja limfocytów T CD8+ jest mniej zależna od tego szlaku kostymulacji. W rzeczywistości, podczas gdy wszystkie komórki CD4+ wykazują ekspresję CD28 na swojej błonie, występuje to tylko w około 50% komórek CD8+.19 Ponadto wykazano, że komórki CD4+ wykazują większą odpowiedź na wiązanie CD2820. Co więcej, promotor CD28 nie jest bezwzględnym wymogiem aktywacji CTL.21 Wszystko to zapewniłoby podwójną korzyść terapeutyczną w praktyce klinicznej. Z jednej strony abatacept działa preferencyjnie na komórkę docelową w patogenezie choroby. Ponadto zmniejszone działanie na aktywność limfocytów CD8+ zapewniłoby lepszy profil bezpieczeństwa w zakresie powikłań wirusowych i nowotworowych.
Aktywacja limfocytów T CD4+ jest punktem wyjścia dla kaskady prozapalnej z produkcją dużej ilości cytokin i proliferacją komórek, która utrwalona i utrzymywana, jak w RZS, prowadzi do bardzo aktywnego przewlekłego zapalenia, zdolnego do niszczenia tkanek, w których jest wyzwalane, głównie stawów w przypadku RZS8 (ryc. 1). Błona maziowa zaczyna się proliferować dzięki komórkom naciekającym z krwi, w tym samym limfocytom T i ich podtypom, a także limfocytom B. Monocyty różnicują się w makrofagi i osteoklasty, a także aktywują chondrocyty stawowe. W tym środowisku powstają duże ilości cytokin prozapalnych, takich jak interleukina (IL)-1, IL-6 oraz czynnik martwicy nowotworów (TNF) i wiele innych. Komórki B produkują również autoprzeciwciała takie jak czynnik reumatoidalny czy przeciwciała przeciwko cytrulinowanemu peptydowi. Wszystkie one prowadzą do zniszczenia nie tylko błony maziowej, ale także leżącej pod nią kości i chrząstki.22
Biotechnologia w leczeniu reumatoidalnego zapalenia stawów
Dzięki wspomnianym badaniom opracowano i skomercjalizowano biotechnologiczną produkcję różnych cząsteczek mających na celu blokowanie określonych celów. Pierwsza generacja charakteryzowała się pojawieniem leków neutralizujących TNF: etanerceptu, infliksymabu i adalimumabu oraz anakinry, która hamuje działanie IL-1. Następnie pojawiły się nowe cząsteczki, takie jak abatacept modulujący kostymulację odpowiedzi immunologicznej, certolizumab i golimumab blokujące TNF, rytuksymab skierowany przeciwko receptorowi CD20 limfocytów B oraz tocilizumab blokujący IL-6.7,23-26
Mimo ogromnego skoku w zakresie skuteczności terapeutycznej, jaki nastąpił dzięki wprowadzeniu tych leków, znaczny odsetek chorych, szacowany na 25% do 40%, nie odpowiada na obecnie dostępne na rynku leki lub preparaty biologiczne albo jest dotknięty występowaniem działań niepożądanych.27-32 Potrzeba poprawy tej sytuacji pozostaje zachętą do poszukiwania i opracowywania nowych cząsteczek ukierunkowanych na regulację różnych celów terapeutycznych, które mogłyby poprawić skuteczność terapeutyczną, tak jak w przypadku abataceptu, który selektywnie moduluje aktywację limfocytów T.33
Abatacept jest konstruktem białkowym wytwarzanym za pomocą technologii rekombinacji DNA w komórkach jajnika chomika.34,35 Cząsteczka ta została zaprojektowana w celu zakłócania regulacji szlaków kostymulacyjnych w komórkach T, które odgrywają ważną rolę w patogenezie różnych chorób autoimmunologicznych, zakażeń, odrzucania przeszczepionych narządów i odporności nowotworowej.36
Abatacept jest stosowany w skojarzeniu z metotreksatem u pacjentów z RZS, u których wystąpiła niewystarczająca odpowiedź lub nietolerancja innych leków z grupy DMARDs, w tym metotreksatu (MTX) lub inhibitora TNF-alfa. W wielostawowym młodzieńczym idiopatycznym zapaleniu stawów jest wskazany u pacjentów w wieku 6 lat lub starszych, u których odpowiedź na inne leki z grupy DMARDs, w tym co najmniej jeden lek neutralizujący TNF, była niewystarczająca.35
Mechanizm działania abataceptu
Abatacept jest selektywnym modulatorem sygnału kostymulacyjnego CD80/86-CD28, a jak wcześniej omówiono jest on niezbędny do aktywacji komórek T Abatacept hamuje aktywację komórek T, selektywnie blokując swoiste wiązanie receptora CD80/CD86 w APC z CD28 na komórce T (ryc. 2).22,37 Ta strategia farmakologiczna ma na celu zahamowanie charakterystycznej dla choroby przyspieszonej odpowiedzi immunologicznej/zapalnej i przywrócenie prawidłowej homeostazy w układzie immunologicznym. W rzeczywistości konkurencja między endogennymi CD28 i CTLA4 o wiązanie z CD80/86 jest fizjologicznym mechanizmem wykorzystywanym do regulacji i, w razie potrzeby, zakończenia prawidłowej odpowiedzi immunologicznej. Abatacept, poprzez blokowanie wiązania CD80/86 z CD28, hamuje przekazywanie drugiego sygnału odpowiedzi immunologicznej, który pośrednio wytwarza negatywny sygnał na aktywację komórek T. Ponadto abatacept prawdopodobnie wywiera większy wpływ na zapobieganie powstawaniu sygnału kostymulującego w komórkach T, inaktywując te już aktywne, które nie są związane z komórkami T CTLA4
Lek wspomagający stosowanie
1. Dlaczego abatacept zaliczany jest do grupy leków immunomodulujących? Zasadniczo dlatego, że powoduje wyczerpanie komórek, zwłaszcza limfocytów T, dzięki działaniu farmakologicznemu polegającemu na tym, że nie blokuje wybiórczo określonej cytokiny, co pozwala uniknąć radykalnej supresji szlaków istotnych dla prawidłowego funkcjonowania odpowiedzi immunologicznej.8
2. W jaki sposób zapobiega wiązaniu regionu Fc cząsteczki z jej receptorem? Region Fc abataceptu jest genetycznie zmodyfikowany tak, że nie wiąże się z receptorami CD16 i CD32, a bardzo słabo z receptorem CD64. Taka konstrukcja pozwala ominąć odpowiedzi komórkowe, w których pośredniczy receptor Fc, takie jak cytotoksyczność komórkowa zależna od przeciwciał (ADCC) i cytotoksyczność zależna od dopełniacza (CDC).18 Obie te reakcje wiążą się z lizą komórek i potencjalnymi działaniami niepożądanymi, które można zaobserwować w przypadku leczenia prolongued38. Dlatego zmodyfikowany fragment IgG1 wydaje się być aktywny, zapobiegając w ten sposób działaniom niepożądanym wynikającym z ADCC.39
3. Przeciwzapalne działanie abataceptu. Abatacept znacząco zmniejsza wiele mediatorów zapalenia u pacjentów z RZS, przywracając je do normy, co wykazano w kilku badaniach klinicznych wykorzystanych podczas badań nad lekiem.
W badaniu fazy II-b, trwającym 1 rok, kontrolowanym placebo, u pacjentów z RZS i niewystarczającą odpowiedzią na MTX, pobrano próbki i zmierzono stężenie wybranych markerów w surowicy w dniach poprzedzających infuzję w celu zbadania wpływu abataceptu na mediatory i cytokiny prozapalne. Grupa pacjentów otrzymywała MTX i abatacept w dawce 10mg/kg, według stałego schematu. Grupa kontrolna natomiast była leczona MTX i placebo. Rok po leczeniu, markery w grupie abataceptu 10mg/kg uległy normalizacji, podczas gdy w grupie placebo pozostały podwyższone (TNF: 7.4 vs 10.3pg/ml; FR: 159 vs 225U/l, sIL-2R: vs 1228.3. 1697,1pg/ml IL-6: 7,3 vs 19,9pg/ml).40
4. Immunogenność. Według danych dotyczących leku tylko u 187 z 3877 (4,8%) chorych na RZS leczonych przez okres do 8 lat abataceptem rozwinęły się w trakcie leczenia przeciwciała przeciwko lekowi.41 Przeciwciała przeciwko abataceptowi oceniano u chorych po odstawieniu leku (>42 dni po podaniu ostatniej dawki) i u 103 z 1888 (5,5%) były seropozytywne. Natomiast w innym badaniu u 2000 pacjentów abataceptu oznaczono przeciwciała i stwierdzono, że abatacept ma niską immunogenność.42,43
5. Abatacept i gruźlica. TNF bierze udział w odpowiedzi zapalnej i immunopatologii gruźlicy (TB). W badaniach in vitro wykazano, że TNF zwiększa aktywność fagocytarną i prątkobójczą makrofagów, natomiast in vivo były zaangażowane w początkowe tworzenie i późniejsze utrzymywanie ziarniniaków, co kontroluje wzrost prątków i ogranicza ich rozprzestrzenianie się. W przewlekłym modelu reaktywacji utajonej gruźlicy u myszy badaliśmy ewolucję zakażenia u myszy leczonych abataceptem w porównaniu z inną grupą leczoną mysim monoklonalnym anty-TNF.42 4 miesiące po zakażeniu myszy C57BL/6 Mycobacterium tuberculosis i po potwierdzeniu, że mają utajone zakażenie gruźlicą, myszy były leczone przez 16 tygodni jedną z dwóch interwencji eksperymentalnych. Po tym czasie wszystkie myszy, którym podawano anty-TNF, umierały z powodu rozsianej gruźlicy, a średni czas przeżycia wynosił 44 dni. Przeciwnie, żadna z myszy leczonych abataceptem nie zmarła.
Podczas gdy stężenie IFN-g w surowicy nie zmieniło się w grupie abataceptu, było podwyższone u myszy z anty-TNF. Wzrost ten przypisano zwiększonej infiltracji CD4+ i CD8+ spowodowanej szerokim rozproszeniem kolonii bakterii.
Więc, podczas gdy myszy leczone terapią anty-TNF wykazywały 100% śmiertelność, abatacept nie zmieniał zdolności myszy do organizowania odpowiedzi zapalnej zdolnej do kontrolowania rozprzestrzeniania się gruźlicy. Jednak nadal nie ma wystarczających danych klinicznych, aby potwierdzić te wyniki u ludzi.
6. Antyresorpcyjne działanie abataceptu na przebudowę kości. Aktywność osteoklastów jest zwiększona w RZS, zarówno w stawie, powodując nadżerki kostne, jak i ogólnoustrojowo, osiągając poziom związany z uogólnioną osteoporozą.44,45
W rzeczywistości w błonie maziowej wykazano zwiększenie stężenia liganda aktywatora receptora czynnika jądrowego NF-kB (RANKL).45,46 Abatacept w sposób zależny od dawki hamuje tworzenie osteoklastów u myszy oraz aktywność osteoklastogenną ocenianą in vitro. Badano to w hodowli osteoklastów myszy na płytkach zębinowych, w której mierzono liczbę dołków resorpcyjnych po 6 dniach dodawania różnych dawek abataceptu.47
Lek znacząco zmniejszał obszar resorpcji kości. Dane te sugerują, że abatacept jest cząsteczką, która wiąże się bezpośrednio z komórkami prekursorowymi osteoklastów, hamując ich różnicowanie. Ten mechanizm mógłby tłumaczyć przeciwerozyjne działanie leku u pacjentów z RZS. Rzeczywiście, u pacjentów leczonych abataceptem stwierdzono tendencję do zmniejszania się stężenia RANK i jego liganda RANKL w mazi stawowej, co wiązało się ze zwiększeniem stężenia osteoprotegeryny.48 Chociaż dokładny mechanizm leżący u podstaw tej obserwacji jest niejasny, wyniki te dobrze korelują z poprawą radiologiczną obserwowaną u pacjentów leczonych abataceptem.
7. Wpływ abataceptu na inne komórki układu odpornościowego. Chociaż APC jest komórką docelową, która wiąże abatacept, a makrofagi również wykazują ekspresję receptorów CD80/86 na swojej powierzchni, istnieje niewiele badań badających działanie leku na aktywność tych komórek. Rzeczywiście, ostatnie badanie in vitro wykazało, że makrofagi wykazywały znaczną ekspresję receptorów CD80/86, a leczenie abataceptem znacznie zmniejszało produkcję cytokin49. Wyniki te sugerują, że mechanizm działania leku może być rozszerzony na regulację linii makrofagów, komórek kluczowych w patogenezie choroby.
Abatacept hamuje również migrację pęcherzykową limfocytów T swoistych dla antygenów i w konsekwencji współpracę pomiędzy limfocytami T i pęcherzykowymi limfocytami B w węźle chłonnym. Odkrycie to zaobserwowano in situ w węzłach chłonnych myszy BALB/c50. Po przetoczeniu takim myszom specyficznych antygenowo, wstępnie stymulowanych limfocytów T, kolejna immunizacja myszy wykazała proliferację komórek T i migrację do obszaru limfocytów B. U myszy, którym podawano abatacept, blokowano proliferację i migrację komórek T, ograniczając ich obecność w większości przypadków do paracortex węzła chłonnego. Tak więc długotrwałe leczenie abataceptem zmniejsza proliferację, mobilność i dystrybucję wewnątrzzwojowych limfocytów pamięci autoantygenów, co może prowadzić do zmniejszenia liczby autoprzeciwciał.
Wnioski dotyczące mechanizmu działania produktu Abatacept
Abatacept jest w pełni humanizowaną konstrukcją białkową, składającą się z zewnątrzkomórkowej domeny ludzkiego antygenu 4 związanego z cytotoksycznymi limfocytami T (CTL4) oraz genetycznie zmodyfikowanego fragmentu regionu Fc IgG1, Lek hamuje aktywację komórek T poprzez selektywne blokowanie swoistego wiązania CD80/CD86 z receptorem CD28, a tym samym hamowanie proliferacji komórek T i odpowiedzi immunologicznej limfocytów B. To działanie farmakologiczne prowadzi do zmniejszenia stężenia mediatorów stanu zapalnego u pacjentów z RZS oraz do bezpiecznej i skutecznej odpowiedzi klinicznej.
Konflikt interesów
Dr Gabriel Herrero-Beaumont otrzymał granty badawcze od firmy Bristol-Myers-Squibb. Dr Santos Castañeda otrzymał granty edukacyjne i badawcze od firm Abbott, MSD i Pfizer.
.