o principal constituinte funcional do glóbulo vermelho, servindo como proteína transportadora de oxigênio; é um tipo de hemoproteína em que cada molécula é um tetrâmero composto de quatro monômeros mantidos juntos por ligações fracas. Consiste em dois pares de cadeias de polipeptídeos, as globinas, cada uma delas com uma molécula heme composta de ferro mais uma molécula de protoporfirina. Símbolo Hb.
O átomo de ferro tem uma valência livre e pode ligar uma molécula de oxigénio. Assim, cada molécula de hemoglobina pode ligar uma molécula de oxigénio. A ligação do oxigénio por um monómero aumenta a afinidade pelo oxigénio das outras no tetrâmero. Isso torna a hemoglobina uma proteína de transporte mais eficiente do que uma proteína monomérica, como a mioglobina.
A hemoglobina oxigenada (oxi-hemoglobina) é de cor vermelho vivo; a hemoglobina não ligada ao oxigênio (desoxi-hemoglobina) é mais escura. Isso explica a cor vermelha brilhante do sangue arterial, no qual a hemoglobina está cerca de 97% saturada com oxigênio. O sangue venoso é mais escuro porque está apenas cerca de 20 a 70 por cento saturado, dependendo da quantidade de oxigénio que está a ser usada pelos tecidos. A afinidade da hemoglobina com o monóxido de carbono é 210 vezes mais forte que a sua afinidade com o oxigénio. O complexo formado (carboxihemoglobina) não pode transportar oxigénio. Assim, o envenenamento por monóxido de carbono resulta em hipoxia e asfixia.
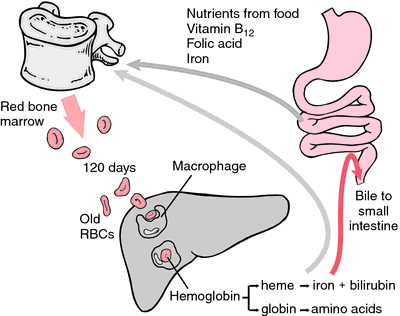
Uma outra forma de hemoglobina que não pode transportar oxigênio é a metemoglobina, na qual o átomo de ferro é oxidado até o estado de oxidação +3. Durante os 120 dias de vida de um glóbulo vermelho, a hemoglobina é oxidada lentamente para a metemoglobina. Pelo menos quatro sistemas enzimáticos diferentes podem converter a metemoglobina de volta em hemoglobina. Quando estes são defeituosos ou sobrecarregados, pode ocorrer metemoglobinemia, com altos níveis de metemoglobina causando dispnéia e cianose.
Uma função secundária da hemoglobina é como parte do sistema tampão do sangue. Os resíduos de histidina nas cadeias de globina atuam como bases fracas para minimizar a mudança no pH do sangue que ocorre à medida que o oxigênio é absorvido e o dióxido de carbono é liberado nos pulmões e à medida que o oxigênio é liberado e o dióxido de carbono é retirado dos tecidos.
As eritrócitos se desgastam ou são danificados, são ingeridos por macrófagos do sistema reticuloendotelial. O anel de porfirina do heme é convertido no pigmento bilirrubina biliar, que é excretado pelo fígado. O ferro é transportado para a medula óssea para ser incorporado à hemoglobina dos eritrócitos recém-formados.
A concentração de hemoglobina no sangue varia com o hematócrito. Os valores normais para a concentração de hemoglobina no sangue são 13,5 a 18,0 g/100 ml em homens e 12,0 a 16,0 g/100 ml em mulheres. A concentração média normal de hemoglobina corpuscular, que é a concentração dentro das hemácias, é de 32 a 36 g/100 ml.
Muitas hemoglobinas anormais decorrentes de mutações foram descobertas. Algumas têm afinidade de oxigênio alterada, algumas são instáveis e em algumas o átomo de ferro é oxidado, resultando em metemoglobinemia congênita. Algumas mutações resultam em uma taxa reduzida de síntese de hemoglobina. Todas essas condições são conhecidas como hemoglobinopatias.
A hemoglobinopatia mais comum é a doença falciforme, causada por uma mutação que substitui o sexto aminoácido da cadeia β, normalmente ácido glutâmico, por valina. A variante hemoglobina α2βS2 é conhecida como Hb S. As mutações que resultam em síntese reduzida de uma das cadeias são chamadas talassemias. Elas podem resultar da deleção do gene para uma cadeia ou de uma mutação no gene regulador que controla a síntese da cadeia.