SONAL DESAI, ARCHITA PATEL ȘI S. Y. GABHE*
C. U. Shah College of Pharmacy, S. N. D. T. Women’s University, Sir Vithaldas Vidya Vihar, Juhu Tara Road, Santacruz (W), Mumbai – 400 049, India
Autor corespondent: S. Y. GABHE E-mail: S. Y. GABHE:
| Date of Accepted | 16-Jan-2011 |
| Date of Revised | 26-Oct-2010 |
| Date of Received | 9-Apr-2009 |
DOI: 10.4103/0250-474X.89762
Abstract
O cromatografie lichidă de înaltă performanță în fază inversă izocratică simplă a fost utilizată pentru a separa trei impurități prezente în proba de 8-cloroteofilină. LC-MS a fost utilizată pentru caracterizarea impurităților. Pe baza datelor din spectrul de masă, structurile acestor impurități au fost caracterizate ca fiind 3,7-dihidro-1,3-dimetil-1H-purină-2,6-dionă (impuritate I), 3,7-dihidro-1,3,7-trimetil-1H-purină-2,6-dionă (impuritate II) și izomer de 8-cloro- 1,3-dimetil-2,6(3H,1H)-purinedionă (impuritate III).
Cuvintele cheie
8-cloroteofilină, impuritate, LC-MS, HPLC cu fază inversă, teofilină
Introducere
Industria medicamentelor în vrac constituie baza tuturor industriilor farmaceutice, deoarece este sursa ingredientelor farmaceutice active (API) de calitate specificată. Provocarea majoră pentru industriile de medicamente în vrac este de a produce medicamentul final de calitatea și puritatea necesară, în mod economic. Puritatea API-urilor depinde de mai mulți factori, cum ar fi materiile prime, metodele de fabricare a acestora și tipul de cristalizare sau procesul de purificare. Cu toate acestea, este aproape imposibil să se obțină materiale absolut pure, deoarece impuritățile se încorporează în ele fie în timpul fabricării, purificării sau depozitării. O impuritate este orice componentă a unei substanțe medicamentoase (cu excepția apei) care nu este o entitate chimică definită ca substanță medicamentoasă. Pentru substanța medicamentoasă produsă prin sinteză chimică, ICH clasifică impuritățile în trei categorii, cum ar fi impuritățile organice, impuritățile anorganice și solvenții reziduali. Impuritățile prezente în substanță pot fi toxice, pot modifica proprietățile fizice sau chimice ale substanței, făcând-o astfel inutilă din punct de vedere medical. Impuritățile pot reduce durata de valabilitate a produsului și pot cauza dificultăți în formulare. Prin urmare, nu numai controlul impurităților, ci și calificarea impurităților este o problemă critică pentru industriile de medicamente în vrac.
Antihistaminicele, cum ar fi dimenhidrinatul și teoclatul de prometazină, sunt medicamente utilizate pe scară largă în tratamentul răului de mișcare. 8-cloroteofilina, din punct de vedere chimic 8-cloro-1,3-dimetil-2,6(1H, 3H)-purinedionă, este un intermediar care este utilizat în prepararea formei de sare a acestor medicamente. Este esențial să se asigure puritatea și siguranța dimenhidrinatului și a teoclatului de prometazină. Pentru a realiza acest lucru, 8-cloroteofilina trebuie să fie obținută cu cea mai mare puritate și cu un profil de impurități cunoscut.
Din analiza literaturii de specialitate se constată că Wadke și colab. au studiat interacțiunile dintre 9-metilizoaloxazină și 3,9-dimetilizoaloxazină cu 8-cloroteofilina. 8-cloroteofilina a fost utilizată ca standard intern pentru determinarea uratului prin metoda HPLC. Acesta a fost, de asemenea, determinat potențiometric. Determinarea simultană a clorhidratului de clorofenoxamină, a 8-cloroteofilinei și a cafeinei în forma farmaceutică multicomponentă a fost realizată printr-o metodă de cromatografie în strat subțire-densitometrică. Gil et al. au investigat comportamentul electroanalitic al 8-cloroteofilinei prin voltametrie ciclică și prin polarografie cu impulsuri diferențiale și au determinat conținutul său în preparate farmaceutice prin polarografie cu impulsuri diferențiale. A fost elaborată și validată o metodă RP-HPLC care indică stabilitatea pentru 8-cloroteofilină împreună cu difenhidramină și cafeină. S-au dezvoltat, de asemenea, metode spectrofotometrice și chimiometrice cu spectru de trecere la zero a spectrului de raport și de trecere la zero a primei derivate pentru determinarea simultană a cafeinei, a 8-cloroteofilinei și a clorhidratului de clorofenoxamină în amestecuri ternare. Până în prezent nu a fost raportată nicio metodă cromatografică și spectroscopică pentru separarea și caracterizarea impurităților prezente în 8-cloroteofilină. Prin urmare, lucrarea de față a fost întreprinsă cu scopul de a izola și caracteriza impuritățile prezente în 8-cloroteofilină folosind tehnici analitice moderne.
8-cloroteofilina a fost un eșantion cadou de la Kores (India) Ltd., Thane. Toate celelalte substanțe chimice și reactivi au fost procurate de la S. D. Fine Chemicals Ltd (Mumbai, India). Solvenții utilizați pentru studiile TLC și TLC pregătitoare au fost de calitate analitică, iar cei utilizați pentru studiile HPLC au fost de calitate HPLC. Pentru prepararea tamponului s-a utilizat acetat de sodiu trihidrat de calitate AR.
Început, s-au efectuat studii TLC pentru a cunoaște numărul de impurități prezente în probă. Ca fază staționară s-au folosit plăci TLC preacoperite cu Silica gel 60GF254 (Merck). Proba a fost dizolvată într-o cantitate minimă de acetat de etil, iar această soluție a fost utilizată pentru pătarea plăcilor TLC. S-au încercat diferite faze mobile. Acetat de etil:toluen:acid acetic glacial (10:0,3:0,5 v/v/v) a prezentat o separare mai bună în comparație cu alte faze mobile. Patru componente au fost separate din proba de 8-cloroteofilină folosind TLC cu valori Rf de 0,029, 0,132, 0,198 și, respectiv, 0,852. 8-cloroteofilina a avut un Rf de 0,852.
După ce a fost dezvoltată faza mobilă pentru TLC, s-a încercat separarea amestecului cu ajutorul TLC pregătitoare. Proba a fost dizolvată în cantitate minimă de acetat de etil și a fost reperată sub formă de bandă. Toate cele trei impurități au fost separate de 8-cloroteofilină prin TLC pregătitoare folosind aceleași condiții cromatografice utilizate în studiile TLC. Diferitele benzi au fost colectate separat și extrase cu acetat de etil. Deoarece cantitățile din fiecare impuritate izolată I, II și III au fost foarte mici, s-a decis izolarea colectivă a acestor impurități. Impuritățile I, II și III au fost denumite în mod colectiv fracțiunea I. Fracțiunea I a fost izolată din 8-cloroteofilină prin TLC pregătitoare. Deoarece fiecare impuritate nu a fost izolată separat, nu au putut fi efectuate diferite tehnici de identificare, cum ar fi IR, RMN. S-a decis să se efectueze investigații suplimentare utilizând LC-MS, care permite separarea și caracterizarea simultană. Înainte de analiza LC-MS, a fost elaborat un profil HPLC pentru fracția I. Pentru studiile HPLC s-a utilizat un cromatograf lichid de înaltă performanță Tosoh echipat cu o pompă alternativă CCPM, un controler de pompă PX8010 și un detector UV. La unitatea de supapă de injecție a fost montată o buclă cu o capacitate de 20 µl. Fracțiunea I a fost dizolvată în acetonitril și a fost supusă analizei HPLC în fază inversă cu o fază mobilă compusă din acetonitril: acetat de sodiu trihidrat (pH 3,57; 0,01 M) (5:95 v/v). Coloana selectată a fost Phenomenex ODS (250×4,6 mm I.D.; dimensiunea particulelor 5 µm). Debitul a fost de 1,5 ml/min, iar detecția a fost monitorizată la o lungime de undă de 280 nm. Faza mobilă a fost filtrată prin filtru de sticlă sinterizată G5 în vid înainte de utilizare și a fost sonicată pentru a elimina bulele de aer. Analiza HPLC a fracțiunii I a evidențiat, de asemenea, trei vârfuri cu timpi de retenție medii de 8,324 min, 14,275 min și, respectiv, 20,933 min (fig. 1). Vârful cu timpul mediu de retenție de 2,086 min a fost vârful solventului.
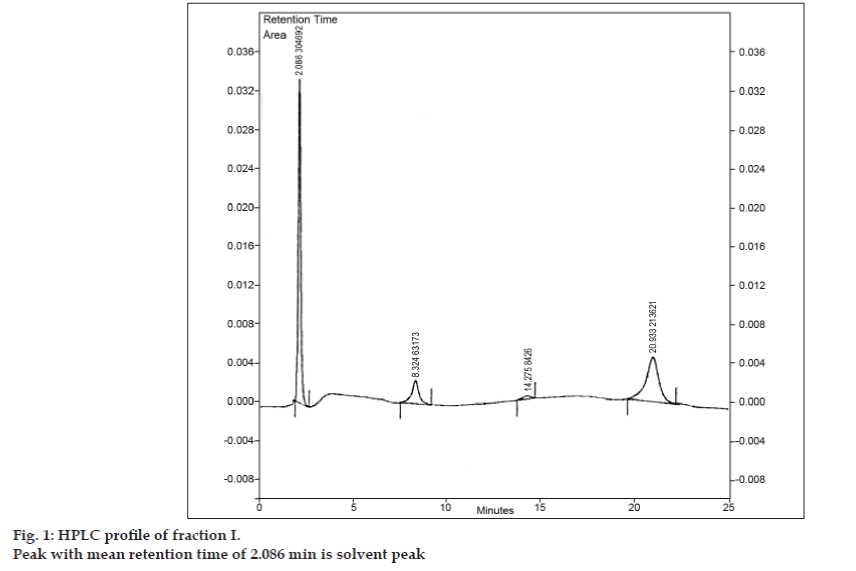
Figura 1: Profilul HPLC al fracției I.
Fracția I a fost apoi supusă analizei LC-MS pentru caracterizarea impurităților. Studiile LC-MS au fost efectuate pe un sistem în care partea LC a constat dintr-un HPLC din seria 1100 (Agilent Technologies, SUA) care cuprinde un degazor în vid (G1322A), o pompă cuaternară (G1311A), un auto-sampler (G1313A) și un detector UV/vizibil (G1314A), iar partea MS a constat dintr-un spectrometru de masă cu triplu cuadrupol Quattro II (Micromass UK Ltd., UK).
În studiile LC-MS, separările cromatografice lichide au fost realizate cu coloana Phenomenex C18 (250×4,6 mm, 5 µm) la temperatura camerei cu faza mobilă de acetonitril: tampon de acetat de sodiu (5:95, v/v) la un debit de 1,5 ml/min. Spectrometrul de masă a funcționat în modul de ionizare prin pulverizare de electroni pozitivi (ESI) cu un raport masă/încărcare (m/z) în intervalul 120-500 m/z. Azotul a fost utilizat ca gaz de nebulizare. Datele au fost achiziționate și prelucrate cu ajutorul software-ului Masslynx. S-au obținut date spectrale de masă ale impurităților (figurile 2, 3 și 4). Calea de fragmentare pentru cele trei vârfuri este caracterizată de pierderea grupului metil și/sau a grupului carbonil. Vârfurile cu valorile m/z de 180,9, 195,7 și 214,9 corespund vârfurilor +, + și, respectiv, +. Conform datelor MS obținute, impuritățile care eluează la 8,324 min, 14,275 min și 20,933 min au fost teofilina (greutate molară 180), cafeina (greutate molară 194) și, respectiv, un izomer de 8-cloroteofilină (greutate molară 214,5) (tabelul 1). Astfel, au fost separate trei impurități, iar structurile lor au fost elucidate pe baza datelor din spectrul de masă (schema 1).
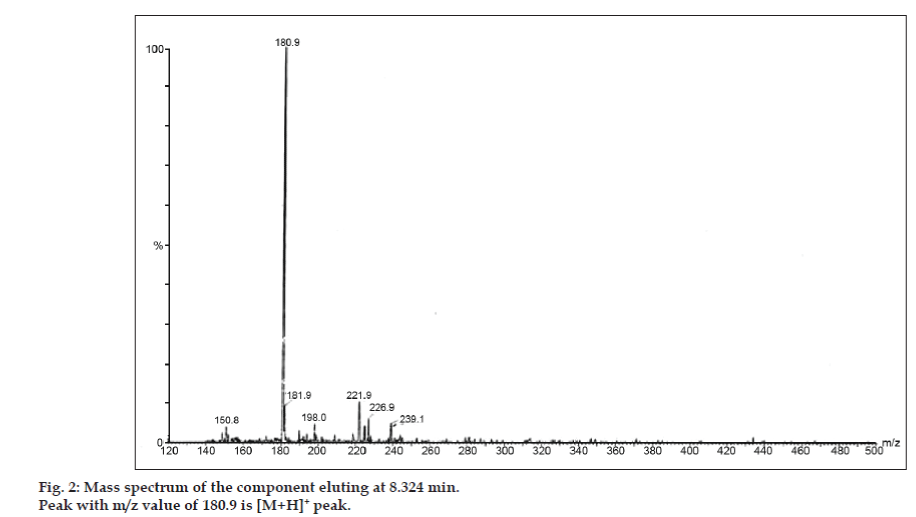
Figură 2: Spectrul de masă al componentei care se eluează la 8,324 min.
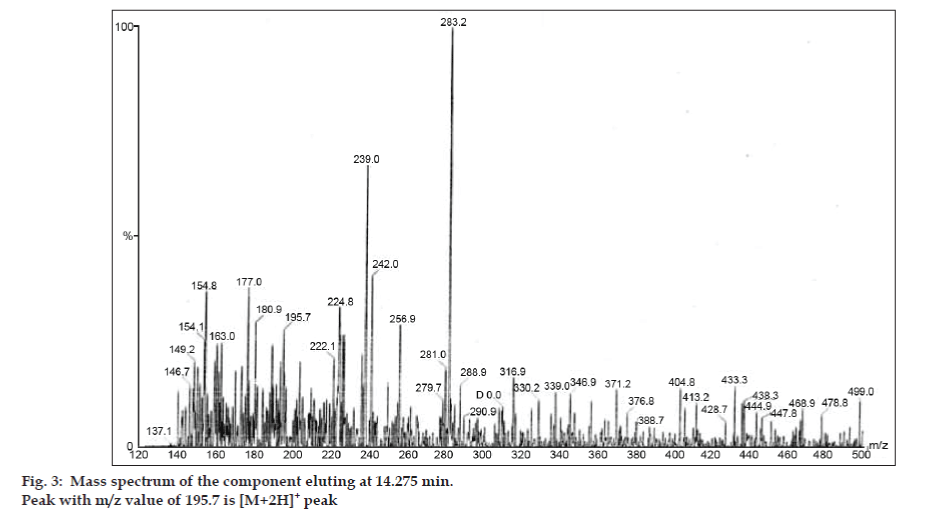
Figură. 3: Spectrul de masă al componentei care eluează la 14,275 min.
Figură
Figură. 4: Spectrul de masă al componentului care eluează la 20,933 min.
| Peak no. |
Timp de retenție timp (min) |
Ioni fragmentari (m/z) | Identificare |
|---|---|---|---|
| 8.324 | 181,9 +, 180,9 +, 150,8 + | Teofilina | |
| 14.275 | 195,7 +, 180,9 +,149,2 +, 137,1+ | Cafeină | |
| 20,933 | 216.9 +, 214,9 +, 171,2 +, 157,7 + | Isomer de 8- Clorozofilină |
TABEL 1: HPLC-MS IDENTIFICAREA FRACȚIUNII I
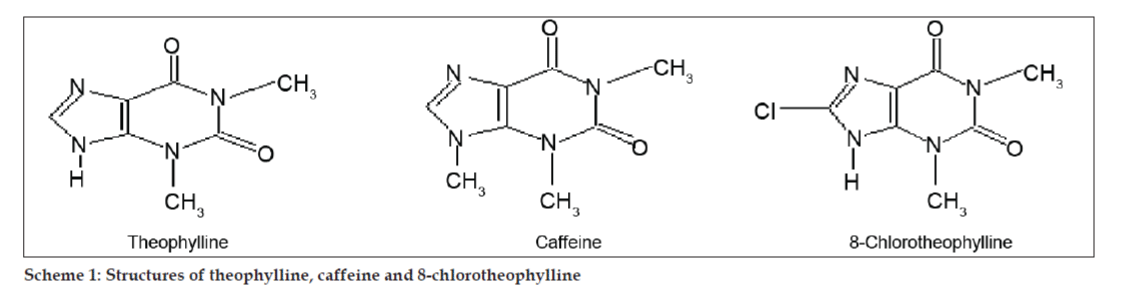
Schema 1: Structurile teofilinei, cafeinei și 8-cloroteofilinei
Recunoștințe
Autorii sunt recunoscători companiei Kores (India) Ltd., Thane pentru că a furnizat o mostră cadou de 8-cloroteofilină.
- Kasture AV, Wadodkar SG, Mahadik KR, More HV. Analiza farmaceutică. Vol. 1. Pune: NiraliPrakashan; 1997. p. 12-4.
- United State Pharmacopoeia, Vol. 26. Rockville, MD: United States Pharmacopoeia Convention, Inc.; 1999. p. 2049-59.
- Ahuja S, Alsante KM. Manual de izolare și caracterizare a impurităților din produsele farmaceutice. California: Academic Press; 2003. p. 6.
- Foye WO, Lemke TL, Williams DA. Principii de chimie medicinală. Ed. a 4-a. New Delhi: B. I. Waverly Pvt Ltd; 1995. p. 419.
- Reynolds JE. Martindale-The Extra Pharmacopoeia. Ediția a 29-a. London: Pharmaceutical Press; 1989. p. 451,459.
- Wadke DA, Guttman DE. Influența formării complexelor asupra vitezei de reacție III. Interacțiunea unor izoalloxazine cu 8-cloroteofilina determinată prin metode spectrale, de solubilitate și cinetice. J Pharm Sci 1965;54:1293.
- Bennett MJ, Patchett BP, Worthy E. O metodă HPLC simplă pentru determinarea uratului în ser și urină folosind 8-cloroteofilina ca standard intern. Med Lab Sci 1984;41:108-11.
- Nikolic K, Medenica M. Potențiometric determination of 8-chlorotheophylline. Microchimica Acta 1986;88:5.
- Bebawy LI, El-Kousy NM. Determinarea simultană a unor forme farmaceutice multicomponente prin metoda densitometrică cantitativă de cromatografie în strat subțire. J Pharm Biomed Anal 1999;20:663-70.
- Gil EP, Blazquez LC, Garcia-MoncoCarra RM, Misiego AS. Polarographic Behavior of 8-Chlorotheophylline and its Determination in Dosage Forms. Electroanalysis 1993;5:343.
- Barbas C, Garcia A, Saavedra L, Castro M. Optimizarea și validarea unei metode de determinare a cafeinei, 8-cloroteofilinei și difenhidraminei prin cromatografie lichidă de înaltă performanță izocratică Test de stres pentru evaluarea stabilității. J ChromatogrA 2000;870:97-103.
- Kelani KM. Determinarea simultană a cafeinei, a clorhidratului de 8-cloroteofilină și a clorhidratului de clorofenoxamină în amestecuri ternare prin metode spectrofotometrice și chimiometrice de trecere la zero a spectrofotometrului de prima derivată și de trecere la zero a spectrului de raport. J AOAC Int 2005;88:1126-34.
.