Introduktion
Rheumatoid artrit (RA) är en autoimmun sjukdom, av polygen karaktär, som kännetecknas av polyartrit med systemiska manifestationer och ökad och allvarlig morbiditet.1,2 RA drabbar 0,5-1 % av befolkningen och orsakar minskad livskvalitet, betydande fysisk funktionsnedsättning och en betydande ekonomisk kostnad.3-6 Det kliniska uttrycket för sjukdomen är varierande och sträcker sig från milda självbegränsande former till en mycket aggressiv, snabb utveckling som kulminerar med förstörelse av den drabbade leden och den därav följande funktionsnedsättningen.7
Genetiska studier har bekräftat förekomsten av ett genetiskt substrat, delvis relaterat till vissa gener som kodar för proteiner som är involverade i T-cellssvar.1 Dessa fynd förstärker betydelsen av den roll som tillskrivs T-cellerna i initieringen och vidmakthållandet av det onormala immunsvaret i denna sjukdom.8
Patogenesen för RA är komplex och involverar olika cellpopulationer som är relaterade till det medfödda och adaptiva immunsvaret. Residensceller i synovium, såsom fibroblastiska synoviocyter B eller makrofager i intima, och inflammatoriska celler från blodet som T-lymfocyter, B-lymfocyter och monocyter9 är involverade i patogenesen. De bidrar alla till den aggressiva omvandlingen av synoviocyt B-fenotypen och utvecklingen av ett intensivt inflammatoriskt infiltrat med slutresultatet brosk- och subchondral benförstörelse10,11 (fig. 1).
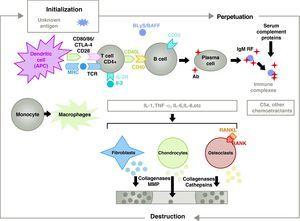
Patofysiologi för reumatoid artrit. Allmän patofysiologisk organisation av reumatoid artrit. AC, antikropp; BAFF, B cell activating factor; BLyS, B lymfocytstimulator; CD, cluster of differentiation; CPA, antigenpresenterande cell; CPH, MHC; CTLA4, lymfocytassocierad antigen 4 T cytotoxisk C5a fraktion komplement 5a, FR, reumatoidfaktor; Ig, immunoglobulin; IL, interleukin; MMP, matrixmetalloproteinaser; RANK, receptoraktivator för kärnfaktorn B kappa; RANKL, receptoraktivatorligand för kärnfaktorn B kappa; RCT, T-cellsreceptor; TNF, tumörnekrosfaktor.
Hej nuvarande behandling av RA bygger på administrering av sjukdomsmodifierande antirheumatiska läkemedel (DMARD) som används ensamma eller i kombination.12 Dessa läkemedel bromsar ledförstöringen, dvs. de kan modifiera sjukdomens naturliga förlopp.4,13 Procentandelen patienter med ett tillfredsställande kliniskt svar är dock låg och kräver ofta tillägg av ett biologiskt läkemedel hos en hög procentandel av patienterna.9,13-15
Under de senaste åren har nya molekyler och terapeutiska mål vars blockering skulle kunna minska eller eliminera det kroniska inflammatoriska svaret identifierats. En av dessa nya molekyler är abatacept. Abatacept är en helt humaniserad proteinkonstruktion som består av den extracellulära domänen av humant cytotoxiskt T-lymfocytassocierat antigen 4 (CTL4) och ett genetiskt modifierat fragment av Fc-regionen av humant immunoglobulin G1 (IgG1), som hämmar de kostimulerande T-cellerna som agerar på immunsvarets egentliga kärna och därför vid sjukdomens uppkomst.
T-cellsaktivering
En effektiv immunaktivering av T-celler kräver deltagande av två grupper av membranreceptorer på antigenpresenterande celler (APC)14 (figurerna 1 och 2). Den första är det fordon som används av APC för att tillhandahålla det tidigare bearbetade specifika antigenet till T-cellen. Trots de enorma ansträngningar som lagts ned på denna forskning kan vi fortfarande inte identifiera de arthritogena antigener som utlöser RA.8 APC:s presentation av ett antigen mot vilket ett specifikt immunsvar utvecklas organiseras genom ett trimolekylärt komplex som består av: molekyler av det stora histokompatibilitetskomplexet (MHC) som finns i APC:n, antigenet mot vilket immunsvaret utvecklas och en membranreceptor på T-cellen (TCR) som är specifik för det antigenet15 (signal- eller signalväg för immunsvaret 1).
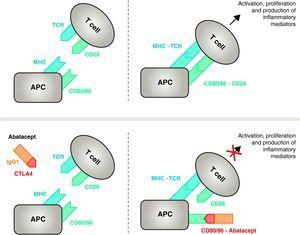
Mekanism för abatacepts verkan. Abataceptfragmentet som omfattar den extracellulära domänen av CTLA4 binder till CD80/CD86-receptorer och förhindrar eller förskjuter dess interaktion med CD28-receptorn. På så sätt blockerar det selektivt den specifika bindningen av CD80/CD86 till CD28-receptorn, vilket patofysiologiskt sett innebär en blockering av den andra signalen för immunaktivering och därmed aktivering av T-celler CPA, antigenpresenterande cell, MHC, större histokompatibilitetskomplex, TCR, T-cellsreceptor.
För att hämma fullständig aktivering kräver T-cellerna en andra uppsättning intercellulära kommunikationsreceptorer mellan APC:s och T-cellerna, som sker genom kostimulatoriska vägar och som utgör den så kallade 2-signal-immunresponsen.14 Även om det finns flera kostimulerande vägar är en viktig, nämligen bindningen av receptorerna CD80 (B7-1)/CD86 (B7-2) på CPA:s membran med CD28-receptorn på T-cellerna.10,16 Samtidig aktivering av båda utlöser intensiv intracellulär signalering i T-cellerna, vilket är nödvändigt för full aktivering, proliferation, överlevnad och cytokinproduktion. 24-48 timmar efter aktiveringen av T-lymfocyter initierar samma intracellulära signalering en regleringsmekanism som syftar till att inaktivera själva responsen. Detta inducerar uttryck av CTLA411 på lymfocytens cellmembran med uppgift att konkurrera med CD28 på grund av dess högre bindningsaffinitet till CD80/CD86.17,18
Aktiveringen av båda undergrupperna av T-celler, CD4+ och CD8+ är beroende av den kostimulerande receptorn CD28. CD4+ T-celler är hjälpande T-celler. De känner igen de peptider som presenteras av MHC-klass II-molekyler som finns på APC. Dessa antigener har sitt ursprung i den exogena väg som bearbetar patogener, t.ex. bakterier. Många autoimmuna sjukdomar är förknippade med ett patologiskt svar från CD4+ T-celler. CD8+ T-celler är å sin sida cytotoxiska lymfocyter (CTL). CD8+ T-celler känner igen antigener, främst virus och tumörer som presenteras av klass I-molekyler MHC. Vid aktivering förmedlar CD8+-cellerna förstörelse av målceller genom produktion av perforin, granzymer och interferon (IFN)-g. Båda subtyperna av T-celler aktiveras genom kostimulering med CD2815, även om aktiveringen av CD8+ T-celler är mindre beroende av denna kostimuleringsväg. Faktum är att medan alla CD4+-celler uttrycker CD28 på sitt membran sker detta endast hos cirka 50 % av CD8+-cellerna.19 Dessutom har det visat sig att CD4+-celler uppvisar ett större svar på CD2820-bindning. Dessutom är CD28-promotorn inte ett absolut krav för aktivering av CTL.21 Allt detta skulle ge en dubbel terapeutisk fördel i klinisk praxis. Å ena sidan verkar abatacept företrädesvis på målcellen i sjukdomens patogenes. Dessutom skulle den minskade verkan på aktiviteten hos CD8+ lymfocyter garantera en bättre säkerhetsprofil när det gäller virala och tumörkomplikationer.
Aktivering av CD4+ T-celler är utgångspunkten för en proinflammatorisk kaskad med produktion av stora mängder cytokiner och cellproliferation som, om den vidmakthålls och upprätthålls, som vid RA, leder till en mycket aktiv kronisk inflammation som kan förstöra de vävnader där den utlöses, oftast lederna vid RA8 (fig. 1). Synoviet börjar proliferera på grund av infiltrerande celler från blodet, inklusive T-lymfocyter själva och deras subtyper samt B-lymfocyter Monocyterna differentieras till makrofager och osteoklaster och aktiverar även ledkörtelns kondrocyter. I denna miljö finns det stora mängder proinflammatoriska cytokiner som interleukin (IL)-1, IL-6 och tumörnekrosfaktor (TNF) och många andra. B-celler producerar också autoantikroppar som reumatoid faktor eller anti-citrullinerade peptidantikroppar. Alla dessa leder till att inte bara synovialmembranet utan även det underliggande benet och brosket förstörs.22
Bioteknik vid behandling av reumatoid artrit
På grund av den ovan nämnda forskningen har bioteknisk tillverkning av olika molekyler som syftar till att blockera specifika mål utvecklats och kommersialiserats. Den första generationen kännetecknades av uppkomsten av TNF-neutraliserande läkemedel: etanercept, infliximab och adalimumab samt anakinra, som hämmar verkan av IL-1. Därefter har nya molekyler dykt upp, t.ex. abatacept för att modulera kostimuleringen av immunsvaret, certolizumab och golimumab för att blockera TNF, rituximab mot CD20-receptorn hos B-lymfocyter och tocilizumab som blockerar IL-6.7,23-26
Trots det enorma språnget i terapeutisk effektivitet till följd av introduktionen av dessa läkemedel svarar en betydande andel patienter, uppskattningsvis mellan 25 och 40 %, inte på de läkemedel eller biologiska preparat som för närvarande saluförs eller drabbas av biverkningar27.Behovet av att förbättra denna situation är fortfarande en uppmuntran för att söka och utveckla nya molekyler som syftar till att reglera olika terapeutiska mål som skulle kunna förbättra den terapeutiska effekten, som till exempel abatacept, som selektivt modulerar aktiveringen av T-celler.33
Abatacept är en proteinkonstruktion som produceras med hjälp av rekombinant DNA-teknik i äggstocksceller från hamster.34,35 Denna molekyl utformades för att störa regleringen av kostimulerande vägar i T-celler, som spelar en viktig roll i patogenesen för olika autoimmuna sjukdomar, infektioner, avstötning av transplanterade organ och tumörimmunitet.36
Abatacept används i kombination med metotrexat hos RA-patienter som har haft otillräckligt svar eller intolerans mot andra DMARDs, inklusive metotrexat (MTX) eller en hämmare av TNF-alfa. Vid polyartikulär juvenil idiopatisk artrit är det indicerat hos patienter som är 6 år eller äldre och som har haft ett otillräckligt svar på andra DMARDs, inklusive minst ett TNF-neutraliserande läkemedel35 .
Abatacepts verkningsmekanism
Abatacept är en selektiv regulator av den kostimulerande CD80/86-CD28-signalen, och som tidigare diskuterats är den väsentlig för aktivering av T-celler Abatacept hämmar aktivering av T-celler genom att selektivt blockera den specifika bindningen av CD80/CD86-receptorn i APC till CD28 på T-cellen (Fig. 22,37 Den farmakologiska strategin syftar till att hämma det accelererade immun/inflammatoriska svaret, som är karakteristiskt för sjukdomen, och återställa normal homeostas i immunsystemet. I själva verket är konkurrensen mellan endogena CD28 och CTLA4 om bindning till CD80/86 den fysiologiska mekanism som används för att reglera och vid behov avsluta ett normalt immunsvar. Abatacept, genom att blockera bindningen av CD80/86 till CD28, hämmar överföringen av en andra signal i immunsvaret, som indirekt ger en negativ signal om T-cellsaktivering. Dessutom har abatacept troligen en större effekt när det gäller att förhindra bildandet av en kostimulerande signal i T-celler, genom att inaktivera de redan aktiva, som inte är bundna till T-cellens CTLA4
Supporting Drug for Use
1. Varför ingår abatacept i gruppen immunmodulerande läkemedel? I grund och botten för att det ger celldepletion, särskilt av T-celler på grund av den farmakologiska verkan som utövas genom att det inte selektivt blockerar ett visst cytokin, vilket undviker ett radikalt undertryckande av viktiga vägar för att immunförsvaret ska fungera korrekt.8
2. Hur förhindrar det att molekylens Fc-region binder sig till sin receptor? Fc-regionen i abatacept är genetiskt modifierad, så att den inte binder till CD16- och CD32-receptorerna och gör det mycket svagt till CD64-receptorn. Denna utformning kringgår cellulära reaktioner som förmedlas av Fc-receptorn, t.ex. antikroppsberoende cellulär cytotoxicitet (ADCC) och komplementberoende cytotoxicitet (CDC).18 Båda är förknippade med celllys, med potentiella negativa effekter som kan ses i förlängda38 behandlingar. Därför verkar det modifierade fragmentet av IgG1 vara aktivt och förhindrar därmed biverkningar till följd av ADCC.39
3. Antiinflammatorisk effekt av abatacept. Abatacept minskar avsevärt många av de inflammatoriska mediatorerna hos patienter med RA och återställer dem till det normala, ett faktum som visats i flera kliniska prövningar som använts när man forskat om läkemedlet.
I en fas II-b, 1-årig, placebokontrollerad studie på patienter med RA och otillräckligt svar på MTX, togs prover och serumnivåerna av utvalda markörer mättes dagarna före infusionen för att studera abatacepts effekt på mediatorer och proinflammatoriska cytokiner. En grupp patienter fick MTX och abatacept 10 mg/kg, enligt regelbundet schema. Kontrollgruppen behandlades under tiden med MTX och placebo. Ett år efter behandlingen hade markörer i abataceptgruppen med 10mg/kg normaliserats, medan de förblev förhöjda i placebogruppen (TNF: 7,4 vs 10,3pg/ml; FR: 159 vs 225U/l, sIL-2R: vs 1228,3. 1697,1pg/ml IL-6: 7,3 vs 19,9pg/ml).40
4. Immunogenicitet. Enligt uppgifter om läkemedlet utvecklade endast 187 av 3877 (4,8 %) patienter med RA som behandlats i upp till 8 år med abatacept antikroppar mot läkemedlet under behandlingen.41 Antikroppar mot abatacept utvärderades hos patienter efter att de slutat med läkemedlet (>42 dagar efter den sista dosen), och hos 103 av 1888 (5,5 %) var seropositiva. I en annan studie av 2000 patienter abatacept mättes däremot antikroppar och man drog slutsatsen att abatacept har låg immunogenicitet.42,43
5. Abatacept och tuberkulos. TNF deltar i det inflammatoriska svaret och immunopatologin vid tuberkulos (TB). In vitro-studier visar att TNF ökar den fagocytiska aktiviteten och mykobakteriedödande makrofagen, medan in vivo var involverade i den initiala bildningen och det efterföljande underhållet av granulom, något som kontrollerar tillväxten av mykobakterier och begränsar dess spridning. I en kronisk modell för reaktivering av latent tuberkulos hos möss studerade vi infektionens utveckling hos möss som behandlades med abatacept jämfört med en annan grupp som behandlades med en murin monoklonal anti-TNF.42 Fyra månader efter att ha infekterat C57BL/6-möss med Mycobacterium tuberculosis och, efter att ha bekräftat att de hade en latent tuberkulosinfektion, behandlades mössen i 16 veckor med en av två experimentella interventioner. Efter denna tid dog alla möss som behandlades med anti-TNF av spridd tuberkulos med en genomsnittlig överlevnad på 44 dagar. Däremot dog ingen av de möss som behandlades med abatacept.
Men medan koncentrationen av IFN-g i serum inte förändrades i abataceptgruppen, var den förhöjd hos mössen med anti-TNF. Denna ökning tillskrevs den ökade infiltrationen av CD4+ och CD8+ som orsakades av den utbredda spridningen av bakteriekolonierna.
Så, medan möss som behandlades med anti-TNF-behandling uppvisade en dödlighet på 100 %, förändrade abatacept inte mössens förmåga att organisera ett inflammatoriskt svar som kunde kontrollera spridningen av tuberkulos. Det finns dock fortfarande inte tillräckligt med kliniska data för att bekräfta dessa resultat hos människor.
6. Antiresorptiv effekt av abatacept på benremodellering. Osteoklastaktiviteten ökar vid RA, både i leden, vilket orsakar benerosioner, och systemiskt, och når nivåer som förknippas med generaliserad osteoporos.44,45
I själva verket har en ökning av ligandreceptoraktivatorn av kärnfaktorn NF-kB (RANKL) påvisats i synovialmembranet.45,46 Abatacept hämmar dosberoende murin osteoklastbildning och den osteoklastogena aktiviteten som bedömts in vitro. Detta studerades i murina osteoklaster odlade på dentinplattor, där man mätte antalet resorptionsgropar efter 6 dagars tillsats av olika doser abatacept.47
Läkemedlet minskade signifikant arean av benresorption. Dessa data tyder på att abatacept är en molekyl som binder direkt till osteoklastprekursorcellerna och hämmar deras differentiering. Denna mekanism skulle kunna förklara läkemedlets antierosiva effekt hos patienter med RA. Faktum är att patienter som behandlades med abatacept uppvisade en minskande trend i nivåerna av RANK och dess ligand RANKL i synovium, allt förknippat med ökat osteoprotegerin.48 Även om den exakta mekanismen som ligger bakom denna observation är oklar, korrelerar dessa fynd väl med den radiologiska förbättring som observerats hos patienter som behandlats med abatacept.
7. Effekter av abatacept i andra immunceller. Även om APC är den målcell som binder abatacept och makrofager också uttrycker CD80/86-receptorer på sin yta, finns det få studier som undersöker läkemedlets verkan på aktiviteten hos dessa celler. En nyligen genomförd in vitro-studie har visat att makrofager uppvisar ett tydligt uttryck för CD80/86-receptorerna och att behandling med abatacept minskar produktionen av cytokiner49 avsevärt. Dessa resultat tyder på att läkemedlets verkningsmekanism skulle kunna utvidgas till att omfatta regleringen av makrofaglinjen, nyckelceller i sjukdomens patogenes.
Abatacept undertrycker också follikulär migration av antigenspecifika T-celler och följaktligen samarbetet mellan T-celler och follikulära B-celler i lymfkörteln. Detta fynd har observerats in situ i lymfkörtlar hos BALB/c50-möss. Efter transfusion av sådana möss med antigenspecifika förstimulerade T-celler visade en efterföljande immunisering av möss T-cellsproliferation och migration till B-lymfocytområdet. Hos möss som behandlades med abatacept blockerades T-cellernas proliferation och migration, vilket begränsade deras förekomst i de flesta fall i lymfkörtelns paracortex. Långvarig behandling med abatacept minskar således proliferation, rörlighet och fördelning av intraganglionära autoantigenminneslymfocyter, vilket skulle kunna leda till en minskning av autoantikroppar.
Slutsatser om Abatacepts verkningsmekanism
Abatacept är en helt humaniserad proteinkonstruktion som består av den extracellulära domänen av humant cytotoxiskt T-lymfocyt-associerat antigen 4 (CTL4) och ett genetiskt modifierat fragment av Fc-regionen av IgG1, utformad för att störa regleringen av kostimulering av T-lymfocyter Läkemedlet hämmar aktivering av T-celler genom att selektivt blockera den specifika bindningen av CD80/CD86 till CD28-receptorn och därför hämmar T-cellsproliferation och immunsvar från B-lymfocyter Denna farmakologiska verkan resulterar i minskade nivåer av inflammatoriska mediatorer hos patienter med RA och i ett säkert och effektivt kliniskt svar.
Intressekonflikter
Dr Gabriel Herrero-Beaumont har fått forskningsbidrag från Bristol-Myers-Squibb. Dr Santos Castañeda har fått utbildnings- och forskningsbidrag från Abbott, MSD och Pfizer.