den viktigaste funktionella beståndsdelen i den röda blodkroppen, som fungerar som syrebärande protein; det är en typ av hemoprotein där varje molekyl är en tetramer som består av fyra monomerer som hålls samman av svaga bindningar. Det består av två par polypeptidkedjor, globinerna, som var och en har en fäst heme-molekyl bestående av järn och en protoporfyrinmolekyl. Symbol Hb.
Järnatomen har en fri valens och kan binda en syremolekyl. Varje hemoglobinmolekyl kan således binda en syremolekyl. När en monomer binder syre ökar affiniteten för syre hos de andra monomerna i tetrameren. Detta gör hemoglobin till ett effektivare transportprotein än ett monomeriskt protein som myoglobin.
Syrehaltigt hemoglobin (oxyhemoglobin) har en ljusröd färg; hemoglobin som inte är bundet till syre (deoxyhemoglobin) är mörkare. Detta förklarar den klarröda färgen på arteriellt blod, där hemoglobinet är cirka 97 procent mättat med syre. Venöst blod är mörkare eftersom det bara är mättat till 20-70 procent, beroende på hur mycket syre vävnaderna använder. Hemoglobinets affinitet för kolmonoxid är 210 gånger starkare än dess affinitet för syre. Det komplex som bildas (karboxyhemoglobin) kan inte transportera syre. Kolmonoxidförgiftning leder således till hypoxi och kvävning.
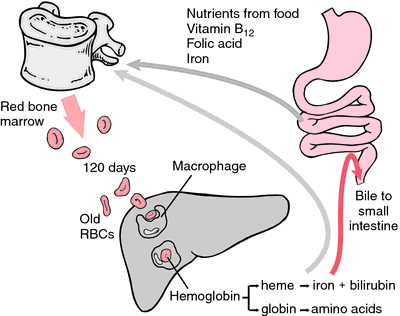
En annan form av hemoglobin som inte kan transportera syre är methemoglobin, där järnatomen är oxiderad till oxidationstillstånd +3. Under de röda blodkropparnas 120 dagars livslängd oxideras hemoglobin långsamt till methemoglobin. Minst fyra olika enzymsystem kan omvandla methemoglobin tillbaka till hemoglobin. När dessa är defekta eller överbelastade kan methemoglobinemi uppstå, med höga methemoglobinnivåer som orsakar dyspné och cyanos.
En sekundär funktion för hemoglobin är som en del av blodets buffertsystem. Histidinresterna i globinkedjorna fungerar som svaga baser för att minimera förändringen i blodets pH-värde som sker när syre absorberas och koldioxid frigörs i lungorna och när syre levereras och koldioxid tas upp från vävnaderna.
När erytrocyter slits ut eller skadas, intas de av makrofager i det retikuloendoteliala systemet. Hems porfyrinring omvandlas till gallpigmentet bilirubin, som utsöndras av levern. Järnet transporteras till benmärgen för att införlivas i hemoglobinet i nybildade erytrocyter.
Hemoglobinkoncentrationen i blodet varierar med hematokriten. Normalvärdena för hemoglobinkoncentrationen i blodet är 13,5 till 18,0 g/100 ml hos män och 12,0 till 16,0 g/100 ml hos kvinnor. Den normala genomsnittliga corpuskulära hemoglobinkoncentrationen, som är koncentrationen i de röda blodkropparna, är 32 till 36 g/100 ml.
Många onormala hemoglobiner som uppstår på grund av mutationer har upptäckts. Vissa har förändrad syreaffinitet, vissa är instabila och i vissa är järnatomen oxiderad, vilket resulterar i medfödd methemoglobinemi. Vissa mutationer resulterar i en minskad hastighet av hemoglobinsyntesen. Alla sådana tillstånd kallas hemoglobinopatier.
Den vanligaste hemoglobinopatien är sicklecellanemi, som orsakas av en mutation som ersätter den sjätte aminosyran i β-kedjan, normalt glutaminsyra, med valin. Varianten hemoglobin α2βS2 är känd som Hb S. Mutationer som resulterar i minskad syntes av en av kedjorna kallas thalassemias. De kan bero på deletion av genen för en kedja eller på en mutation i den regulatoriska genen som styr syntesen av kedjan.